E16.5 mouse dentate gyrus
library(COTAN)
library(data.table)
library(Matrix)
library(ggrepel)
#> Loading required package: ggplot2
library(latex2exp)mycolours <- c(A = "#8491B4B2", B = "#E64B35FF")
my_theme = theme(axis.text.x = element_text(size = 14,
angle = 0, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.text.y = element_text(size = 14,
angle = 0, hjust = 0, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.title.x = element_text(size = 14,
angle = 0, hjust = 0.5, vjust = 0,
face = "plain", colour = "#3C5488FF"),
axis.title.y = element_text(size = 14,
angle = 90, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"))data = as.data.frame(fread(paste(data_dir,
"separation_age/E16.5.csv", sep = "/")))
data = as.data.frame(data)
rownames(data) = data$V1
data = data[, 2:ncol(data)]
data[1:10, 1:10]
#> X10X82_1_CAGAACAAGTTCTG. X10X82_1_TCACTTCTCGCATC.
#> 0610007P14Rik 4 1
#> 0610009B22Rik 1 1
#> 0610009L18Rik 0 0
#> 0610009O20Rik 2 0
#> 0610010F05Rik 0 1
#> 0610010K14Rik 4 0
#> 0610011F06Rik 0 0
#> 0610012D04Rik 0 0
#> 0610012G03Rik 4 1
#> 0610025J13Rik 0 0
#> X10X82_1_TCACTCATATGCTG. X10X82_1_ATGGCTCACGCGGT.
#> 0610007P14Rik 1 1
#> 0610009B22Rik 0 1
#> 0610009L18Rik 0 1
#> 0610009O20Rik 0 0
#> 0610010F05Rik 0 0
#> 0610010K14Rik 0 1
#> 0610011F06Rik 1 1
#> 0610012D04Rik 0 0
#> 0610012G03Rik 3 2
#> 0610025J13Rik 0 0
#> X10X82_1_GGTGCCAACGCACC. X10X82_1_TGAGTTCAAGCCTA.
#> 0610007P14Rik 3 1
#> 0610009B22Rik 1 1
#> 0610009L18Rik 0 0
#> 0610009O20Rik 0 0
#> 0610010F05Rik 0 1
#> 0610010K14Rik 0 0
#> 0610011F06Rik 1 0
#> 0610012D04Rik 0 0
#> 0610012G03Rik 1 2
#> 0610025J13Rik 0 0
#> X10X82_1_CACGATCGCGTTTC. X10X82_1_TATGCGTCCGCTGA.
#> 0610007P14Rik 2 0
#> 0610009B22Rik 0 2
#> 0610009L18Rik 0 0
#> 0610009O20Rik 0 0
#> 0610010F05Rik 1 0
#> 0610010K14Rik 0 0
#> 0610011F06Rik 1 0
#> 0610012D04Rik 0 0
#> 0610012G03Rik 1 3
#> 0610025J13Rik 1 0
#> X10X82_1_TGCTATCGGAAACG. X10X82_1_GTTAGAGCGCTTAT.
#> 0610007P14Rik 2 4
#> 0610009B22Rik 0 0
#> 0610009L18Rik 0 0
#> 0610009O20Rik 0 0
#> 0610010F05Rik 0 0
#> 0610010K14Rik 0 1
#> 0610011F06Rik 0 1
#> 0610012D04Rik 0 0
#> 0610012G03Rik 1 2
#> 0610025J13Rik 0 0Define a directory where the ouput will be stored.
0.1 Analytical pipeline
Initialize the COTAN object with the row count table and the metadata for the experiment.
obj = new("scCOTAN", raw = data)
obj = initRaw(obj, GEO = "GSE104323", sc.method = "10X",
cond = "mouse dentate gyrus E16.5")
#> [1] "Initializing S4 object"Now we can start the cleaning. Analysis requires and starts from a matrix of raw UMI counts after removing possible cell doublets or multiplets and low quality or dying cells (with too high mtRNA percentage, easily done with Seurat or other tools).
If we do not want to consider the mitochondrial genes we can remove them before starting the analysis.
genes_to_rem = rownames(obj@raw[grep("^mt",
rownames(obj@raw)), ]) #genes to remove : mithocondrial
obj@raw = obj@raw[!rownames(obj@raw) %in%
genes_to_rem, ]
cells_to_rem = colnames(obj@raw[which(colSums(obj@raw) ==
0)])
obj@raw = obj@raw[, !colnames(obj@raw) %in%
cells_to_rem]We want also to define a prefix to identify the sample.
0.2 Data cleaning
First we create the directory to store all information regardin the data cleaning.
if (!file.exists(out_dir)) {
dir.create(file.path(out_dir))
}
if (!file.exists(paste(out_dir, "cleaning",
sep = ""))) {
dir.create(file.path(out_dir, "cleaning"))
}ttm = clean(obj)
#> [1] "Start estimation mu with linear method"
#> [1] 14131 2285
#> rowname X10X82_1_TACTGAGGCATGGT.
#> 9692 Ptgds 910.01759
#> 7119 Malat1 144.82597
#> 8232 Nnat 56.04377
#> 4619 Ftl1 52.71443
#> 1127 Apoe 44.94599
#> 1404 Atp1a2 43.83621
#> 2343 Cdkn1c 38.84221
#> 2840 Cox4i1 32.73844
#> 2854 Cox8a 32.73844
#> 10515 Rps29 32.18355
#> 3746 Eef1a1 28.29933
#> 10510 Rps27 27.18955
#> 13168 Uqcr11 25.52488
#> 10458 Rpl41 24.41511
#> 10454 Rpl37a 23.30533
obj = ttm$object
ttm$pca.cell.2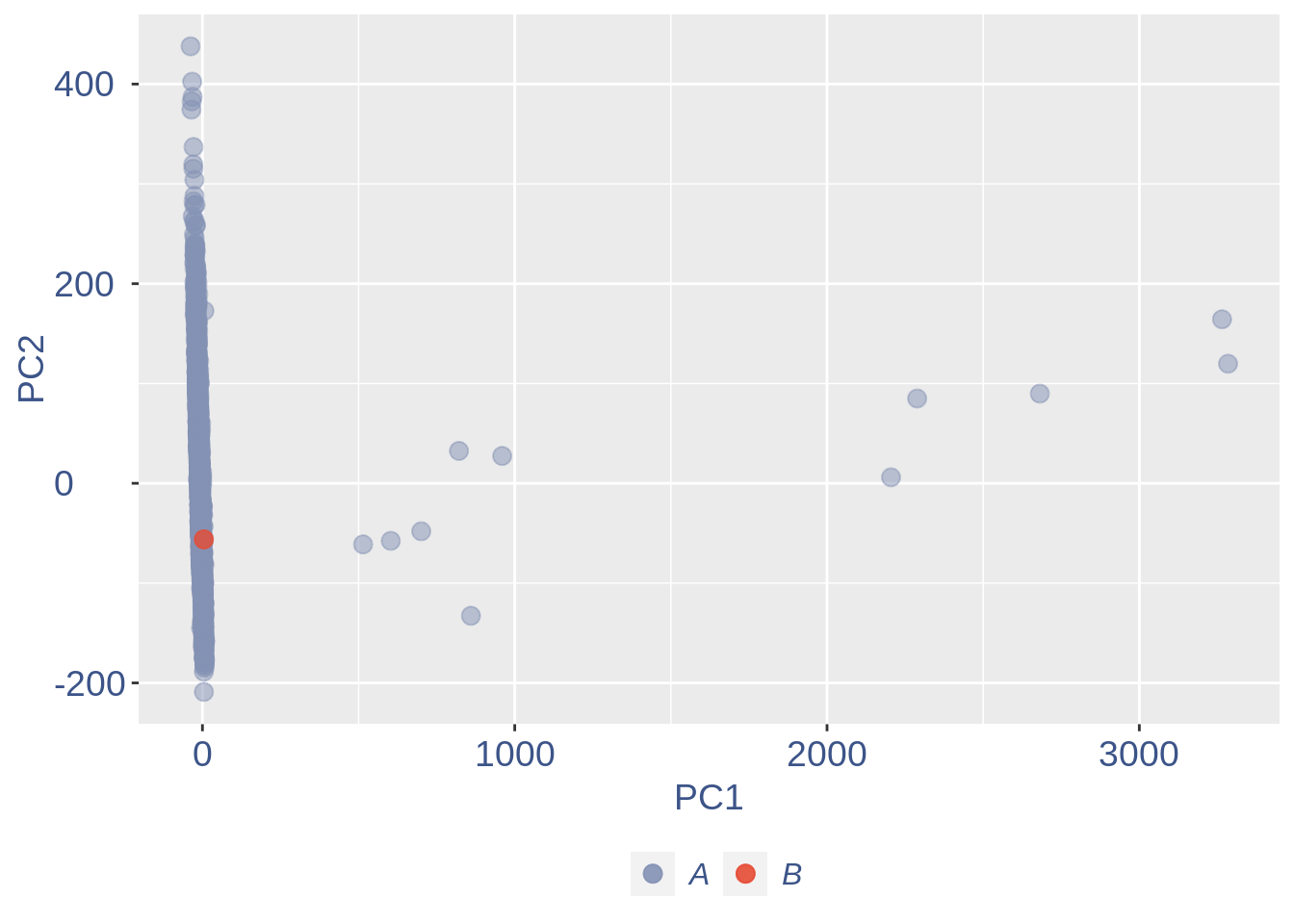
Run this when B cells need to be removed.
pdf(paste(out_dir, "cleaning/", t, "_", n_it,
"_plots_before_cells_exlusion.pdf", sep = ""))
ttm$pca.cell.2
ggplot(ttm$D, aes(x = n, y = means)) + geom_point() +
geom_text_repel(data = subset(ttm$D,
n > (max(ttm$D$n) - 15)), aes(n,
means, label = rownames(ttm$D[ttm$D$n >
(max(ttm$D$n) - 15), ])), nudge_y = 0.05,
nudge_x = 0.05, direction = "x",
angle = 90, vjust = 0, segment.size = 0.2) +
ggtitle("B cell group genes mean expression") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF",
size = 20, face = "italic", vjust = -5,
hjust = 0.02))
dev.off()
#> png
#> 2
if (length(ttm$cl1) < length(ttm$cl2)) {
to_rem = ttm$cl1
} else {
to_rem = ttm$cl2
}
n_it = n_it + 1
obj@raw = obj@raw[, !colnames(obj@raw) %in%
to_rem]
# obj@raw = obj@raw[rownames(obj@raw)
# %in% rownames(cells),colnames(obj@raw)
# %in% colnames(cells)]
gc()
#> used (Mb) gc trigger (Mb) max used (Mb)
#> Ncells 3706915 198.0 6015681 321.3 6015681 321.3
#> Vcells 231648395 1767.4 347181239 2648.8 347179504 2648.8
ttm = clean(obj)
#> [1] "Start estimation mu with linear method"
#> [1] 14126 2284
#> rowname X10X82_1_GTTCCGTTCGGGCT. X10X82_1_CGTAAGTAGAAAGG.
#> 12935 Ttr 729.43369 1156.30913
#> 7116 Malat1 140.43227 68.94445
#> 10455 Rpl41 60.65046 85.11661
#> 4618 Ftl1 38.26270 31.06756
#> 10502 Rps23 24.83005 41.28156
#> 10512 Rps29 29.30760 34.04664
#> 10507 Rps27 23.20185 28.93965
#> 10489 Rps14 29.30760 26.38615
#> 10451 Rpl37a 25.23710 33.19548
#> 1792 Bsg 21.57365 24.25823
#> 4617 Fth1 22.79480 22.13032
#> 10416 Rpl13 26.45825 18.30007
#> 3745 Eef1a1 19.94545 20.42799
#> 10450 Rpl37 22.38775 20.85357
#> 10424 Rpl18a 21.98070 19.57682
#> X10X82_1_CGGATAGCCACGCT.
#> 12935 891.76077
#> 7116 123.00149
#> 10455 62.52575
#> 4618 43.56303
#> 10502 37.92546
#> 10512 28.70035
#> 10507 29.21285
#> 10489 25.62531
#> 10451 21.01275
#> 1792 25.62531
#> 4617 25.62531
#> 10416 21.52526
#> 3745 25.62531
#> 10450 22.03777
#> 10424 21.52526
# ttm = clean.sqrt(obj, cells)
obj = ttm$object
ttm$pca.cell.2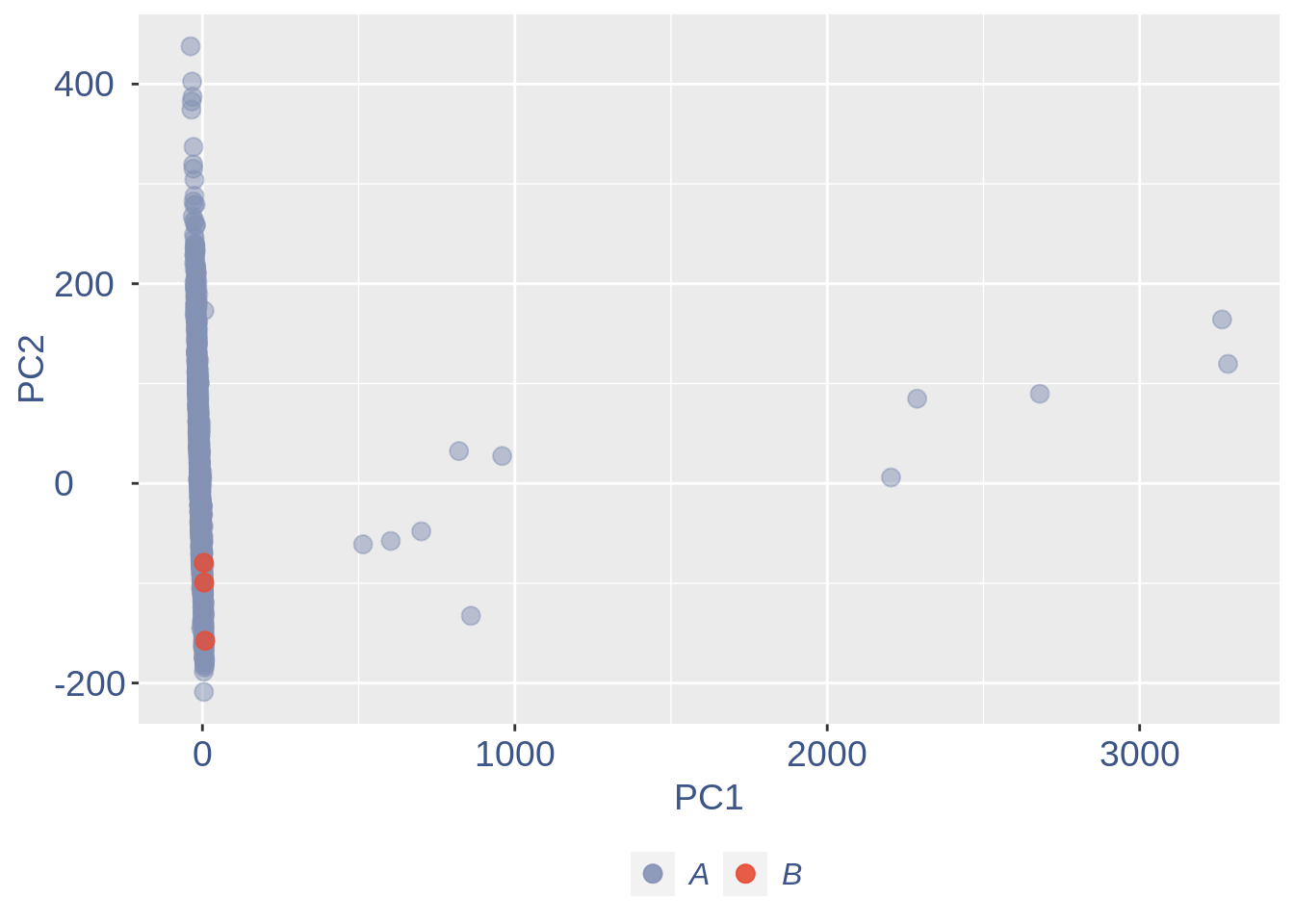
Run this only in the last iteration, instead the previous code, when B cells group has not to be removed.
pdf(paste(out_dir, "cleaning/", t, "_", n_it,
"_plots_before_cells_exlusion.pdf", sep = ""))
ttm$pca.cell.2
ggplot(ttm$D, aes(x = n, y = means)) + geom_point() +
geom_text_repel(data = subset(ttm$D,
n > (max(ttm$D$n) - 15)), aes(n,
means, label = rownames(ttm$D[ttm$D$n >
(max(ttm$D$n) - 15), ])), nudge_y = 0.05,
nudge_x = 0.05, direction = "x",
angle = 90, vjust = 0, segment.size = 0.2) +
ggtitle(label = "B cell group genes mean expression",
subtitle = " - B group NOT removed -") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF",
size = 20, face = "italic", vjust = -10,
hjust = 0.02), plot.subtitle = element_text(color = "darkred",
vjust = -15, hjust = 0.01))
dev.off()
#> png
#> 2To colour the pca based on nu_j (so the cells’ efficiency).
nu_est = round(obj@nu, digits = 7)
plot.nu <- ggplot(ttm$pca_cells, aes(x = PC1,
y = PC2, colour = log(nu_est)))
plot.nu = plot.nu + geom_point(size = 1,
alpha = 0.8) + scale_color_gradient2(low = "#E64B35B2",
mid = "#4DBBD5B2", high = "#3C5488B2",
midpoint = log(mean(nu_est)), name = TeX(" $ln (\\nu) $ ")) +
ggtitle("Cells PCA coloured by cells efficiency") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF",
size = 20), legend.title = element_text(color = "#3C5488FF",
size = 14, face = "italic"), legend.text = element_text(color = "#3C5488FF",
size = 11), legend.key.width = unit(2,
"mm"), legend.position = "right")
pdf(paste(out_dir, "cleaning/", t, "_plots_PCA_efficiency_colored.pdf",
sep = ""))
plot.nu
dev.off()
#> png
#> 2
plot.nu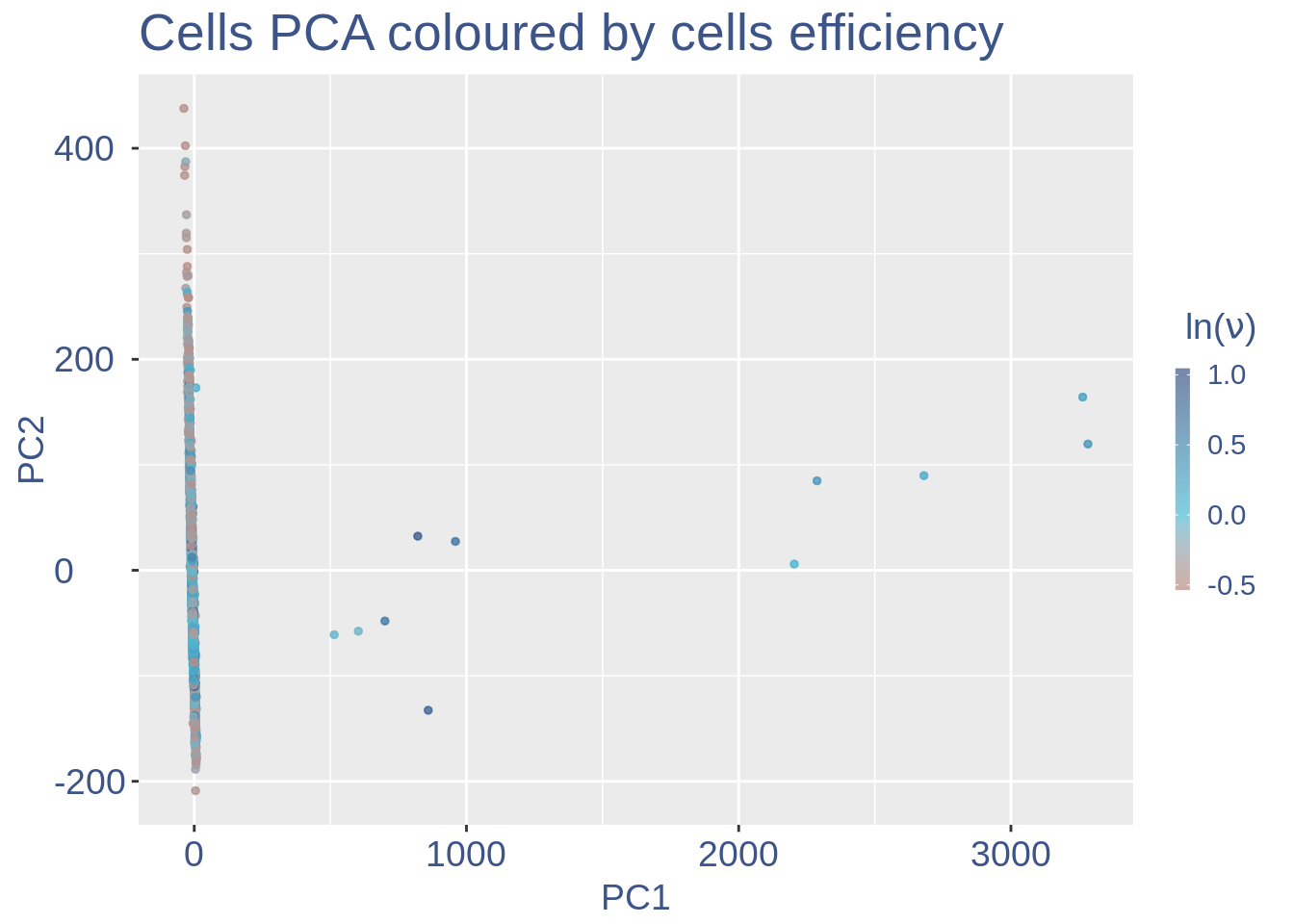
The next part is used to remove the cells with efficiency too low.
nu_df = data.frame(nu = sort(obj@nu), n = c(1:length(obj@nu)))
ggplot(nu_df, aes(x = n, y = nu)) + geom_point(colour = "#8491B4B2",
size = 1) + my_theme #+ ylim(0,1) + xlim(0,70)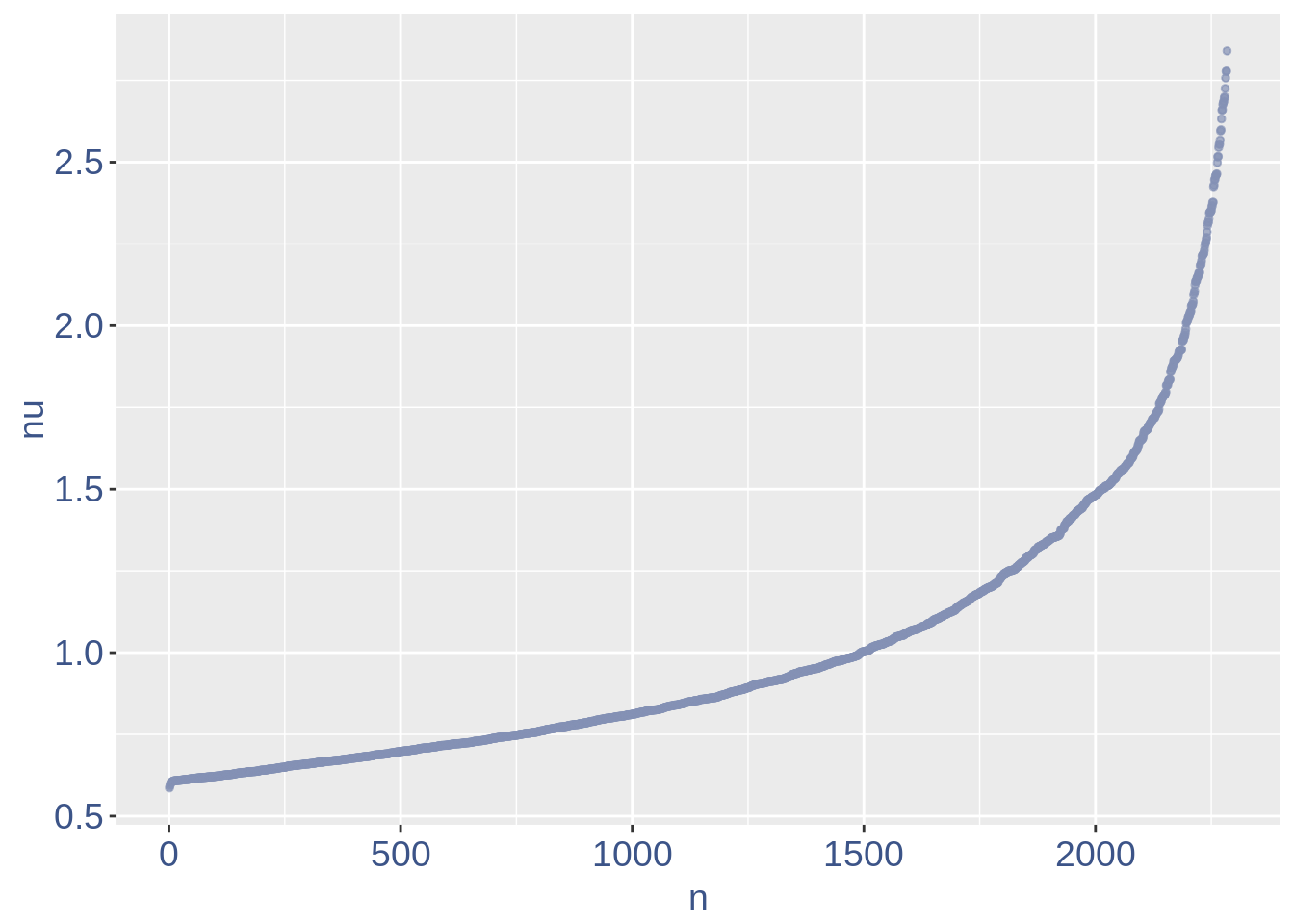
We can zoom on the smallest values and, if we detect a clear elbow, we can decide to remove the cells.
yset = 0.61 #threshold to remove low UDE cells
plot.ude <- ggplot(nu_df, aes(x = n, y = nu)) +
geom_point(colour = "#8491B4B2", size = 1) +
my_theme + ylim(0.5, 1) + xlim(0, 800) +
geom_hline(yintercept = yset, linetype = "dashed",
color = "darkred") + annotate(geom = "text",
x = 200, y = 0.5, label = paste("to remove cells with nu < ",
yset, sep = " "), color = "darkred",
size = 4.5)
pdf(paste(out_dir, "cleaning/", t, "_plots_efficiency.pdf",
sep = ""))
plot.ude
dev.off()
#> png
#> 2
plot.ude
We also save the defined threshold in the metadata and re-run the estimation.
obj@meta[(nrow(obj@meta) + 1), 1:2] = c("Threshold low UDE cells:",
yset)
to_rem = rownames(nu_df[which(nu_df$nu <
yset), ])
obj@raw = obj@raw[, !colnames(obj@raw) %in%
to_rem]Repeat the estimation after the cells are removed.
ttm = clean(obj)
#> [1] "Start estimation mu with linear method"
#> [1] 14119 2259
#> rowname X10X82_1_GTTCCGTTCGGGCT. X10X82_1_CGTAAGTAGAAAGG.
#> 12928 Ttr 732.65805 1161.42367
#> 7114 Malat1 141.05303 69.24941
#> 10450 Rpl41 60.91855 85.49309
#> 4616 Ftl1 38.43184 31.20498
#> 10497 Rps23 24.93981 41.46415
#> 10507 Rps29 29.43715 34.19724
#> 10502 Rps27 23.30441 29.06765
#> 10484 Rps14 29.43715 26.50286
#> 10446 Rpl37a 25.34866 33.34231
#> 1790 Bsg 21.66902 24.36553
#> 4615 Fth1 22.89556 22.22820
#> 10411 Rpl13 26.57521 18.38101
#> 3743 Eef1a1 20.03362 20.51834
#> 10445 Rpl37 22.48671 20.94581
#> 10419 Rpl18a 22.07787 19.66341
#> X10X82_1_CGGATAGCCACGCT.
#> 12928 895.71685
#> 7114 123.54715
#> 10450 62.80314
#> 4616 43.75628
#> 10497 38.09371
#> 10507 28.82767
#> 10502 29.34245
#> 10484 25.73899
#> 10446 21.10597
#> 1790 25.73899
#> 4615 25.73899
#> 10411 21.62075
#> 3743 25.73899
#> 10445 22.13553
#> 10419 21.62075
obj = ttm$object
ttm$pca.cell.2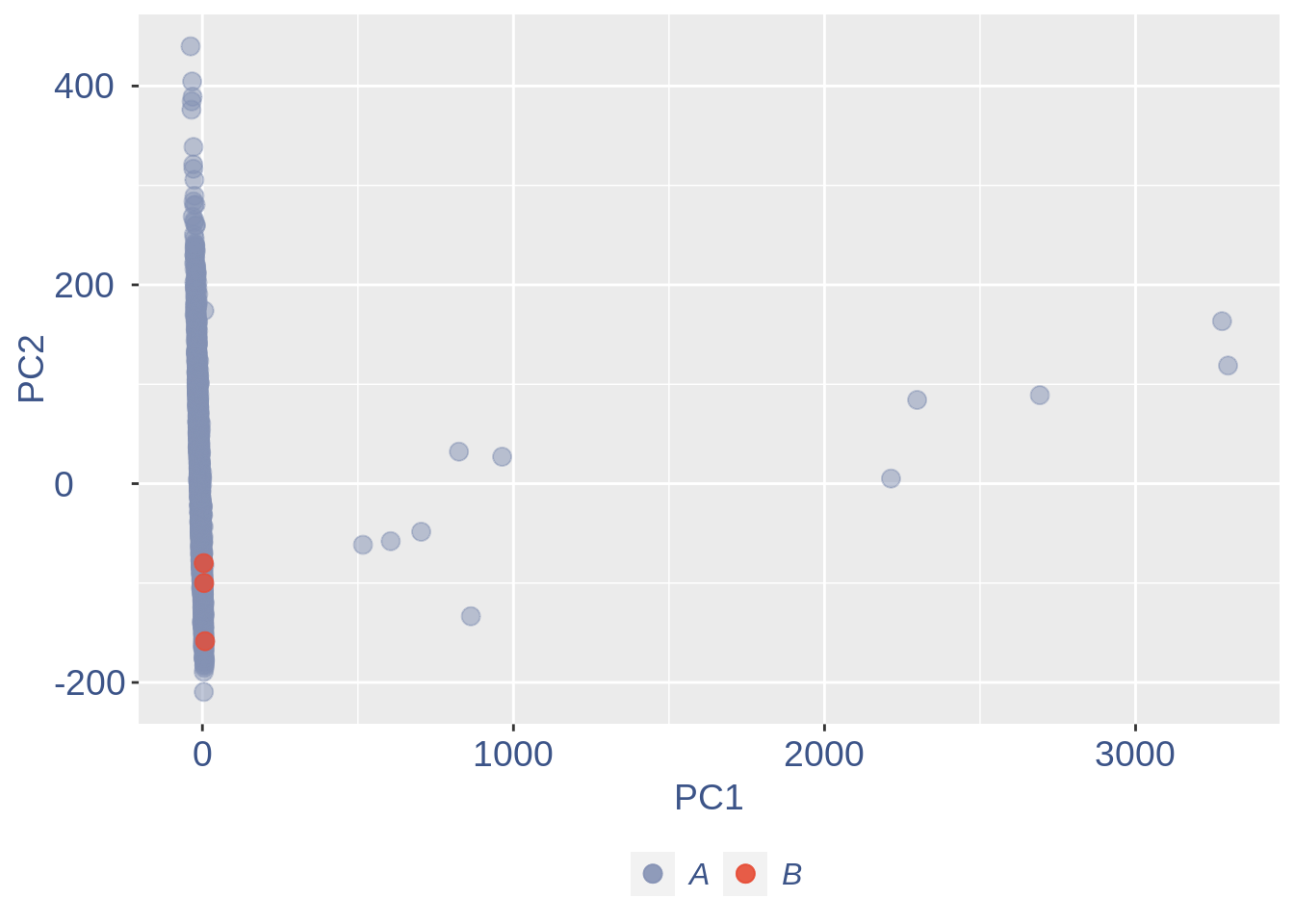
In case we do not want to remove anything, we can run:
pdf(paste(out_dir,"cleaning/",t,"_plots_efficiency.pdf", sep = ""))
ggplot(nu_df, aes(x = n, y=nu)) + geom_point(colour = "#8491B4B2", size=1) +my_theme + #xlim(0,100)+
annotate(geom="text", x=50, y=0.25, label="nothing to remove ", color="darkred")
dev.off()
#> png
#> 2Just to check again, we plot the final efficiency coloured PCA.
nu_est = round(obj@nu, digits = 7)
plot.nu <- ggplot(ttm$pca_cells, aes(x = PC1,
y = PC2, colour = log(nu_est)))
plot.nu = plot.nu + geom_point(size = 2,
alpha = 0.8) + scale_color_gradient2(low = "#E64B35B2",
mid = "#4DBBD5B2", high = "#3C5488B2",
midpoint = log(mean(nu_est)), name = TeX(" $ln (\\nu) $ ")) +
ggtitle("Cells PCA coloured by cells efficiency: last") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF",
size = 20), legend.title = element_text(color = "#3C5488FF",
size = 14, face = "italic"), legend.text = element_text(color = "#3C5488FF",
size = 11), legend.key.width = unit(2,
"mm"), legend.position = "right")
pdf(paste(out_dir, "cleaning/", t, "_plots_PCA_efficiency_colored_FINAL.pdf",
sep = ""))
plot.nu
dev.off()
#> png
#> 2
plot.nu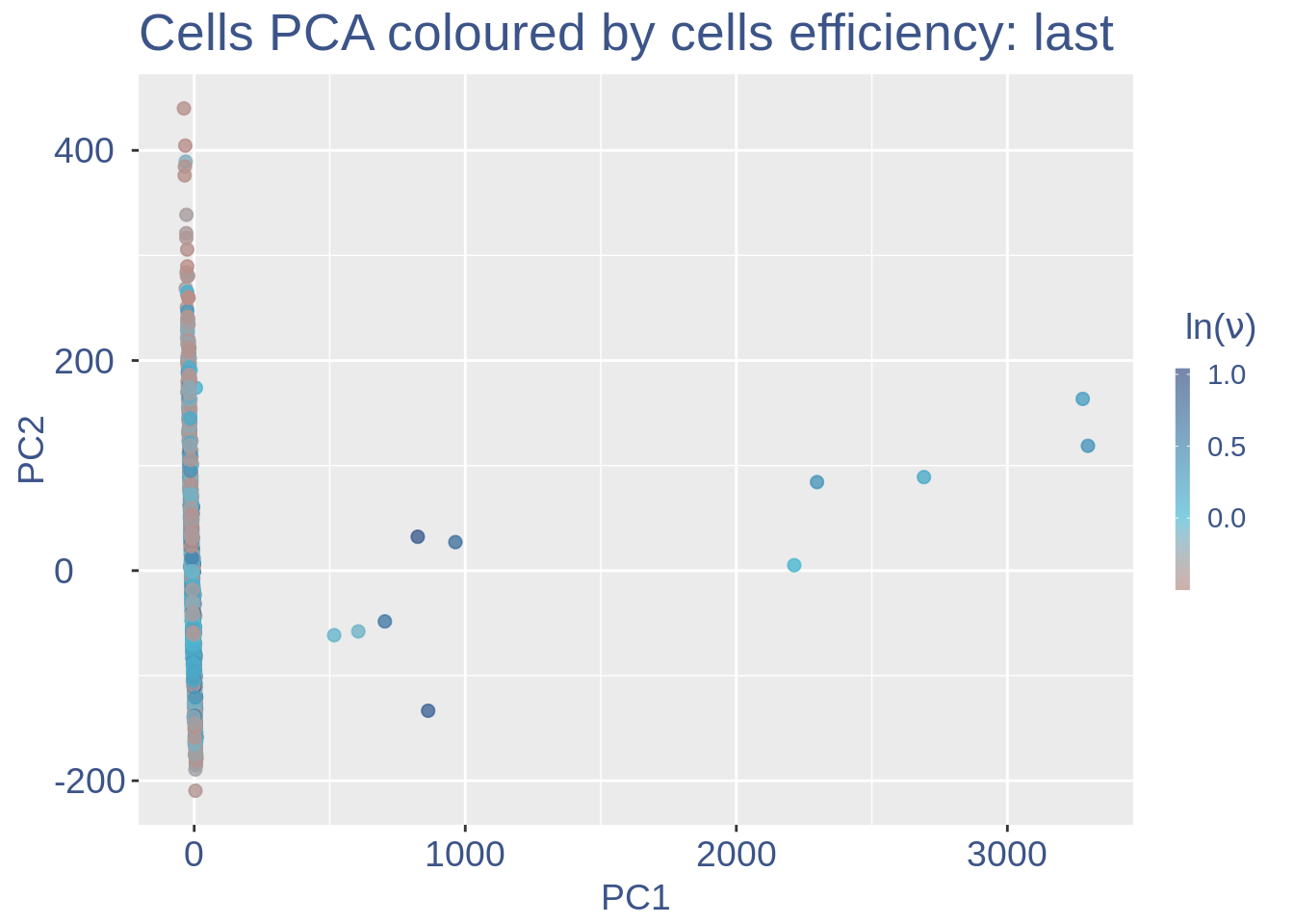
COTAN analysis: in this part all the contingency tables are computed and used to get the statistics (S) To storage efficiency of all the observed tables only the yes_yes is stored.
Note that if will be necessary re-computing the yes-yes table, the yes-yes table should be cancelled before running cotan_analysis.
obj = cotan_analysis(obj)
#> [1] "cotan analysis"
#> [1] "mu estimator creation"
#> [1] "save effective constitutive genes"
#> [1] "start a minimization"
#> [1] "Next gene: Btk number 1810"
#> [1] "Next gene: Eif2d number 3820"
#> [1] "Next gene: Hdhd2 number 5830"
#> [1] "Next gene: Myl12b number 7840"
#> [1] "Next gene: Rab22a number 9850"
#> [1] "Next gene: Swi5 number 11860"
#> [1] "Next gene: Zfp74 number 13870"
#> [1] "Final trance!"
#> [1] "a min: -0.15966796875 | a max 871.25 | negative a %: 23.9152084222507"COEX evaluation and storing.
saveRDS(obj, file = paste(out_dir, t, ".cotan.RDS",
sep = ""))
obj = get.coex(obj)
#> [1] "coex dataframe creation"
#> [1] "Generating contingency tables for observed data"
#> [1] "creation of observed yes/yes contingency table"
#> [1] "mu estimator creation"
#> [1] "expected contingency tables creation"
#> [1] "The distance between estimated n of zeros and observed number of zero is 0.0638973580200531 over 14058"
#> [1] "Done"
#> [1] "coex estimation"
#> [1] "Diagonal coex values substituted with 0"
# saving the structure
saveRDS(obj, file = paste(out_dir, t, ".cotan.RDS",
sep = ""))Automatic data elaboration (without cleaning)
obj2 = automatic.COTAN.object.creation(df = data,
GEO = "GSE104323", sc.method = "10X",
cond = "E16_dent_gy", out_dir = "Data/")
#> [1] "Initializing S4 object"
#> [1] "Condition E16_dent_gy"
#> [1] "n cells 2285"
#> [1] "Start estimation mu with linear method"
#> [1] 14131 2285
#> rowname X10X82_1_TACTGAGGCATGGT.
#> 9692 Ptgds 910.01759
#> 7119 Malat1 144.82597
#> 8232 Nnat 56.04377
#> 4619 Ftl1 52.71443
#> 1127 Apoe 44.94599
#> 1404 Atp1a2 43.83621
#> 2343 Cdkn1c 38.84221
#> 2840 Cox4i1 32.73844
#> 2854 Cox8a 32.73844
#> 10515 Rps29 32.18355
#> 3746 Eef1a1 28.29933
#> 10510 Rps27 27.18955
#> 13168 Uqcr11 25.52488
#> 10458 Rpl41 24.41511
#> 10454 Rpl37a 23.30533
#> [1] "Cotan analysis function started"
#> [1] "cotan analysis"
#> [1] "mu estimator creation"
#> [1] "save effective constitutive genes"
#> [1] "start a minimization"
#> [1] "Next gene: Btg1 number 1810"
#> [1] "Next gene: Eif2b3 number 3820"
#> [1] "Next gene: Hddc3 number 5830"
#> [1] "Next gene: Myh2 number 7840"
#> [1] "Next gene: Rab14 number 9850"
#> [1] "Next gene: Suv420h1 number 11860"
#> [1] "Next gene: Zfp706 number 13870"
#> [1] "a min: -0.16796875 | a max 881.25 | negative a %: 23.9800995024876"
#> [1] "Only analysis time 2.71626739501953"
#> [1] "Cotan coex estimation started"
#> [1] "coex dataframe creation"
#> [1] "Generating contingency tables for observed data"
#> [1] "creation of observed yes/yes contingency table"
#> [1] "mu estimator creation"
#> [1] "expected contingency tables creation"
#> [1] "The distance between estimated n of zeros and observed number of zero is 0.0633983950493963 over 14070"
#> [1] "Done"
#> [1] "coex estimation"
#> [1] "Diagonal coex values substituted with 0"
#> [1] "Total time 5.0609365383784"
#> [1] "Only coex time 1.99707407156626"
#> [1] "Saving elaborated data locally at Data/E16_dent_gy.cotan.RDS"1 Analysis of the elaborated data
In this case the test possible macroscopic differences between cleaned and not cleaned data.
The initial cell number was:
obj@meta
#> V1 V2
#> 1 GEO: GSE104323
#> 2 scRNAseq method: 10X
#> 3 starting n. of cells: 2285
#> 4 Condition sample: mouse dentate gyrus E16.5
#> 5 Threshold low UDE cells: 0.61While the final number of cells was:
So were removed:
cells
1.1 GDI with cleaning
quant.p = get.GDI(obj)
#> [1] "function to generate GDI dataframe"
#> [1] "Using S"
#> [1] "function to generate S "
head(quant.p)
#> sum.raw.norm GDI exp.cells
#> 0610007P14Rik 7.771356 1.757084 61.000443
#> 0610009B22Rik 7.481643 1.433632 52.058433
#> 0610009L18Rik 5.796126 1.570076 13.103143
#> 0610009O20Rik 5.366675 1.915944 8.632138
#> 0610010F05Rik 7.335761 2.961452 44.001771
#> 0610010K14Rik 7.414252 1.500532 49.535193In the third column of this data frame is reported the percentage of cells expressing the gene.
NPGs = c("Nes", "Vim", "Sox2", "Sox1", "Notch1",
"Hes1", "Hes5", "Pax6") #,'Hes3'
PNGs = c("Map2", "Tubb3", "Neurod1", "Nefm",
"Nefl", "Dcx", "Tbr1")
hk = c("Calm1", "Cox6b1", "Ppia", "Rpl18",
"Cox7c", "Erh", "H3f3a", "Taf1", "Taf2",
"Gapdh", "Actb", "Golph3", "Mtmr12",
"Zfr", "Sub1", "Tars", "Amacr")
text.size = 12
quant.p$highlight = with(quant.p, ifelse(rownames(quant.p) %in%
NPGs, "NPGs", ifelse(rownames(quant.p) %in%
hk, "Constitutive", ifelse(rownames(quant.p) %in%
PNGs, "PNGs", "normal"))))
textdf <- quant.p[rownames(quant.p) %in%
c(NPGs, hk, PNGs), ]
mycolours <- c(Constitutive = "#00A087FF",
NPGs = "#E64B35FF", PNGs = "#F39B7FFF",
normal = "#8491B4B2")
f1 = ggplot(subset(quant.p, highlight ==
"normal"), aes(x = sum.raw.norm, y = GDI)) +
geom_point(alpha = 0.1, color = "#8491B4B2",
size = 2.5)
GDI_plot = f1 + geom_point(data = subset(quant.p,
highlight != "normal"), aes(x = sum.raw.norm,
y = GDI, colour = highlight), size = 2.5,
alpha = 0.8) + geom_hline(yintercept = quantile(quant.p$GDI)[4],
linetype = "dashed", color = "darkblue") +
geom_hline(yintercept = quantile(quant.p$GDI)[3],
linetype = "dashed", color = "darkblue") +
geom_hline(yintercept = 1.5, linetype = "dotted",
color = "red", size = 0.5) + scale_color_manual("Status",
values = mycolours) + scale_fill_manual("Status",
values = mycolours) + xlab("log normalized counts") +
ylab("GDI") + geom_label_repel(data = textdf,
aes(x = sum.raw.norm, y = GDI, label = rownames(textdf),
fill = highlight), label.size = NA,
alpha = 0.5, direction = "both", na.rm = TRUE,
seed = 1234) + geom_label_repel(data = textdf,
aes(x = sum.raw.norm, y = GDI, label = rownames(textdf)),
label.size = NA, segment.color = "black",
segment.size = 0.5, direction = "both",
alpha = 0.8, na.rm = TRUE, fill = NA,
seed = 1234) + theme(axis.text.x = element_text(size = text.size,
angle = 0, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.text.y = element_text(size = text.size,
angle = 0, hjust = 0, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.title.x = element_text(size = text.size,
angle = 0, hjust = 0.5, vjust = 0,
face = "plain", colour = "#3C5488FF"),
axis.title.y = element_text(size = text.size,
angle = 90, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
legend.title = element_blank(), legend.text = element_text(color = "#3C5488FF",
face = "italic"), legend.position = "bottom") # titl)
legend <- cowplot::get_legend(GDI_plot)
GDI_plot = GDI_plot + theme(legend.position = "none")
GDI_plot
1.2 GDI without cleaning
quant.p = get.GDI(obj2)
#> [1] "function to generate GDI dataframe"
#> [1] "Using S"
#> [1] "function to generate S "
head(quant.p)
#> sum.raw.norm GDI exp.cells
#> 0610007P14Rik 7.777981 1.756868 60.831510
#> 0610009B22Rik 7.491839 1.442903 51.903720
#> 0610009L18Rik 5.812074 1.560409 13.129103
#> 0610009O20Rik 5.362669 1.918659 8.533917
#> 0610010F05Rik 7.345695 2.959066 43.938731
#> 0610010K14Rik 7.420519 1.505370 49.409190In the third column of this data frame is reported the percentage of cells expressing the gene.
NPGs = c("Nes", "Vim", "Sox2", "Sox1", "Notch1",
"Hes1", "Hes5", "Pax6") #,'Hes3'
PNGs = c("Map2", "Tubb3", "Neurod1", "Nefm",
"Nefl", "Dcx", "Tbr1")
hk = c("Calm1", "Cox6b1", "Ppia", "Rpl18",
"Cox7c", "Erh", "H3f3a", "Taf1", "Taf2",
"Gapdh", "Actb", "Golph3", "Mtmr12",
"Zfr", "Sub1", "Tars", "Amacr")
text.size = 12
quant.p$highlight = with(quant.p, ifelse(rownames(quant.p) %in%
NPGs, "NPGs", ifelse(rownames(quant.p) %in%
hk, "Constitutive", ifelse(rownames(quant.p) %in%
PNGs, "PNGs", "normal"))))
textdf <- quant.p[rownames(quant.p) %in%
c(NPGs, hk, PNGs), ]
mycolours <- c(Constitutive = "#00A087FF",
NPGs = "#E64B35FF", PNGs = "#F39B7FFF",
normal = "#8491B4B2")
f1 = ggplot(subset(quant.p, highlight ==
"normal"), aes(x = sum.raw.norm, y = GDI)) +
geom_point(alpha = 0.1, color = "#8491B4B2",
size = 2.5)
GDI_plot = f1 + geom_point(data = subset(quant.p,
highlight != "normal"), aes(x = sum.raw.norm,
y = GDI, colour = highlight), size = 2.5,
alpha = 0.8) + geom_hline(yintercept = quantile(quant.p$GDI)[4],
linetype = "dashed", color = "darkblue") +
geom_hline(yintercept = quantile(quant.p$GDI)[3],
linetype = "dashed", color = "darkblue") +
geom_hline(yintercept = 1.5, linetype = "dotted",
color = "red", size = 0.5) + scale_color_manual("Status",
values = mycolours) + scale_fill_manual("Status",
values = mycolours) + xlab("log normalized counts") +
ylab("GDI") + geom_label_repel(data = textdf,
aes(x = sum.raw.norm, y = GDI, label = rownames(textdf),
fill = highlight), label.size = NA,
alpha = 0.5, direction = "both", na.rm = TRUE,
seed = 1234) + geom_label_repel(data = textdf,
aes(x = sum.raw.norm, y = GDI, label = rownames(textdf)),
label.size = NA, segment.color = "black",
segment.size = 0.5, direction = "both",
alpha = 0.8, na.rm = TRUE, fill = NA,
seed = 1234) + theme(axis.text.x = element_text(size = text.size,
angle = 0, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.text.y = element_text(size = text.size,
angle = 0, hjust = 0, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.title.x = element_text(size = text.size,
angle = 0, hjust = 0.5, vjust = 0,
face = "plain", colour = "#3C5488FF"),
axis.title.y = element_text(size = text.size,
angle = 90, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
legend.title = element_blank(), legend.text = element_text(color = "#3C5488FF",
face = "italic"), legend.position = "bottom") # titl)
legend <- cowplot::get_legend(GDI_plot)
GDI_plot = GDI_plot + theme(legend.position = "none")
GDI_plot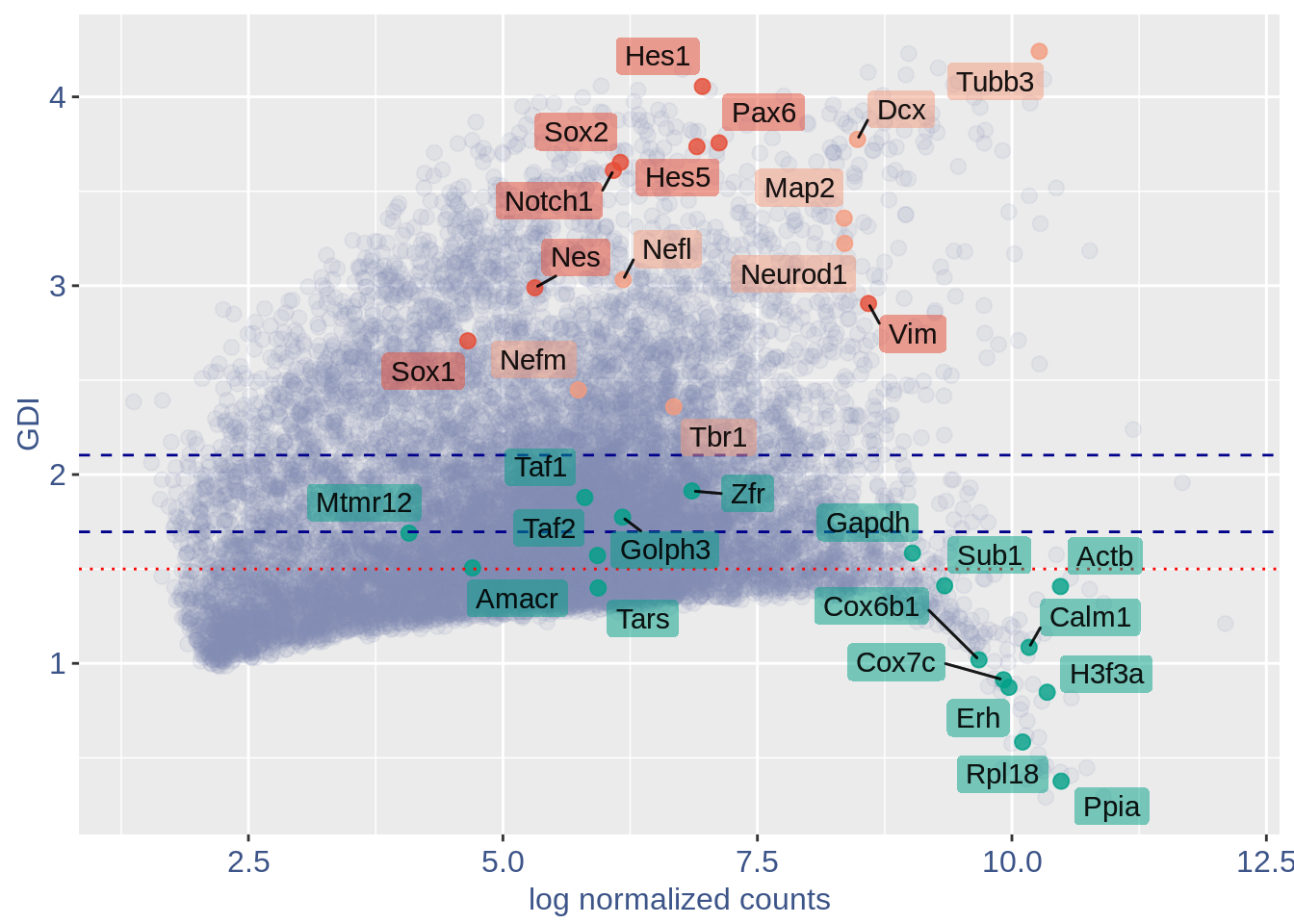
1.3 Heatmaps with cleaning
For the Gene Pair Analysis, we can plot an heatmap with the coex values between two genes sets. To do so we need to define, in a list, the different gene sets (list.genes). Then in the function parameter sets we can decide which sets need to be considered (in the example from 1 to 3). In the condition parameter we should insert an array with each file name prefix to be considered (in the example, the file names is “E15_cortex_clean”).
list.genes = list(Ref.col = PNGs, NPGs = NPGs,
Const. = hk)
plot_heatmap(df_genes = list.genes, sets = c(1:3),
conditions = "E16.5_cleaned", dir = "Data/")
#> [1] "plot heatmap"
#> [1] "Loading condition E16.5_cleaned"
#> [1] "Map2" "Tubb3" "Neurod1" "Nefm" "Nefl" "Dcx" "Tbr1"
#> [1] "Get p-values on a set of genes on columns on a set of genes on rows"
#> [1] "Using function S"
#> [1] "function to generate S "
#> [1] "Ref.col"
#> [1] "NPGs"
#> [1] "Const."
#> [1] "min coex: -0.0118943380956698 max coex 0.010004681362927"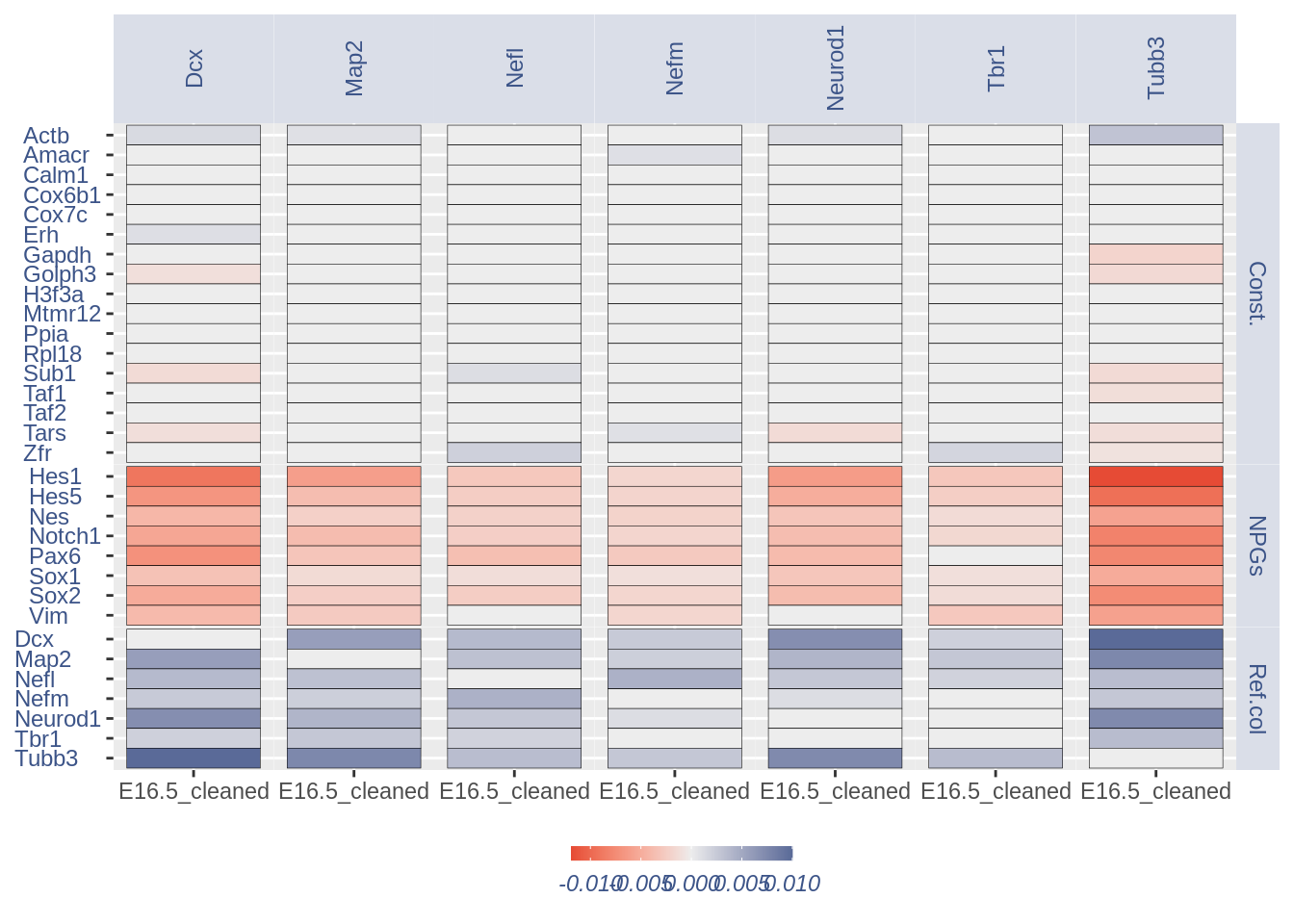
1.4 Heatmaps without cleaning
list.genes = list(Ref.col = PNGs, NPGs = NPGs,
Const. = hk)
plot_heatmap(df_genes = list.genes, sets = c(1:3),
conditions = "E16_dent_gy", dir = "Data/")
#> [1] "plot heatmap"
#> [1] "Loading condition E16_dent_gy"
#> [1] "Map2" "Tubb3" "Neurod1" "Nefm" "Nefl" "Dcx" "Tbr1"
#> [1] "Get p-values on a set of genes on columns on a set of genes on rows"
#> [1] "Using function S"
#> [1] "function to generate S "
#> [1] "Ref.col"
#> [1] "NPGs"
#> [1] "Const."
#> [1] "min coex: -0.0118691182192925 max coex 0.0099495109519959"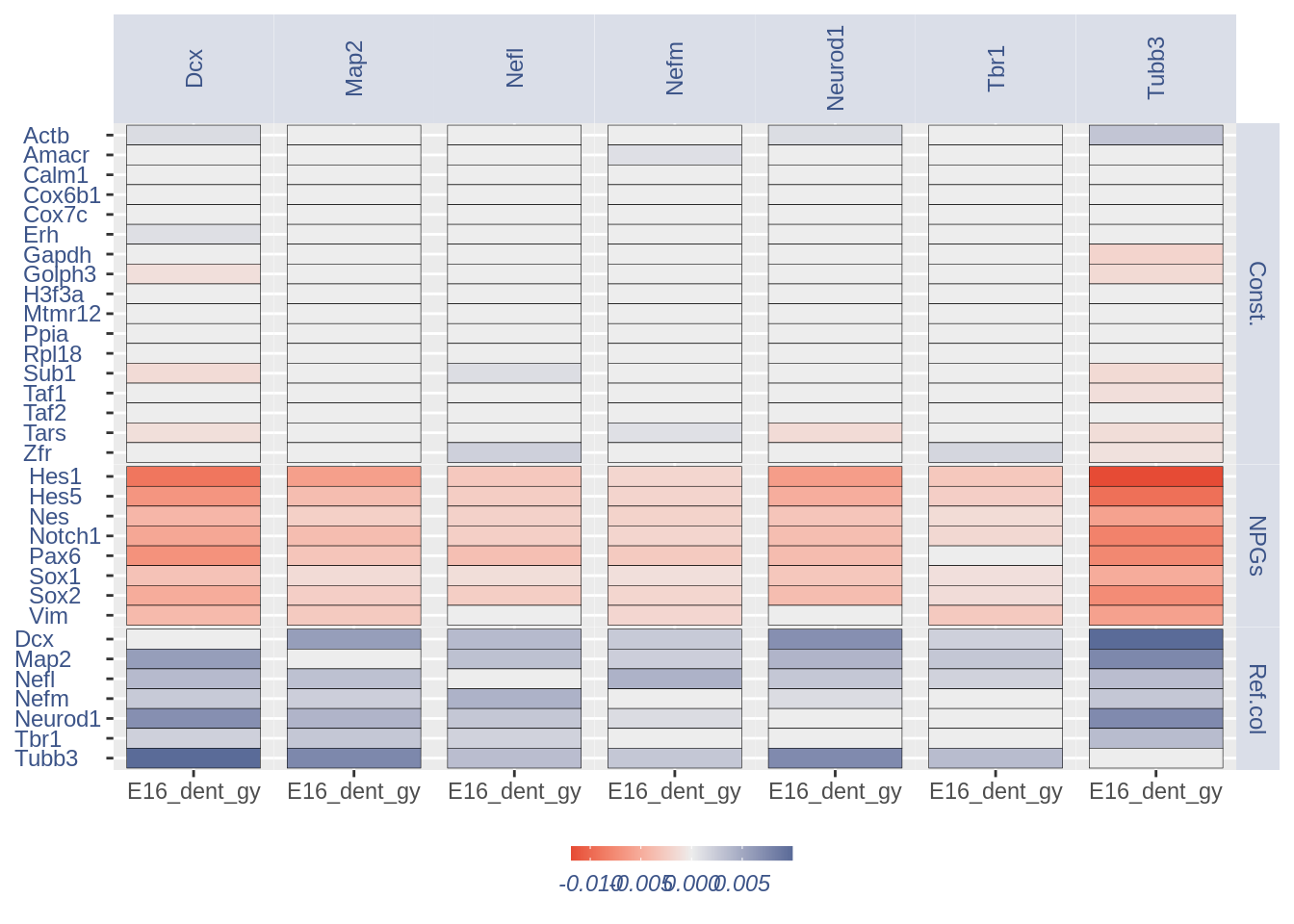
print(sessionInfo())
#> R version 4.0.4 (2021-02-15)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 18.04.5 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/libopenblasp-r0.2.20.so
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] latex2exp_0.5.0 ggrepel_0.9.1 ggplot2_3.3.3 Matrix_1.3-2
#> [5] data.table_1.14.0 COTAN_0.1.0
#>
#> loaded via a namespace (and not attached):
#> [1] Rcpp_1.0.6 lattice_0.20-41 circlize_0.4.12
#> [4] tidyr_1.1.2 png_0.1-7 assertthat_0.2.1
#> [7] digest_0.6.27 utf8_1.2.1 R6_2.5.0
#> [10] stats4_4.0.4 evaluate_0.14 highr_0.8
#> [13] pillar_1.5.1 basilisk_1.2.1 GlobalOptions_0.1.2
#> [16] rlang_0.4.10 jquerylib_0.1.3 S4Vectors_0.28.1
#> [19] GetoptLong_1.0.5 reticulate_1.18 rmarkdown_2.7
#> [22] labeling_0.4.2 stringr_1.4.0 munsell_0.5.0
#> [25] compiler_4.0.4 xfun_0.22 pkgconfig_2.0.3
#> [28] BiocGenerics_0.36.0 shape_1.4.5 htmltools_0.5.1.1
#> [31] tidyselect_1.1.0 tibble_3.1.0 IRanges_2.24.1
#> [34] matrixStats_0.58.0 fansi_0.4.2 withr_2.4.1
#> [37] crayon_1.4.0 dplyr_1.0.4 rappdirs_0.3.3
#> [40] basilisk.utils_1.2.2 grid_4.0.4 gtable_0.3.0
#> [43] jsonlite_1.7.2 lifecycle_1.0.0 DBI_1.1.1
#> [46] magrittr_2.0.1 formatR_1.8 scales_1.1.1
#> [49] stringi_1.5.3 farver_2.1.0 bslib_0.2.4
#> [52] ellipsis_0.3.1 filelock_1.0.2 generics_0.1.0
#> [55] vctrs_0.3.6 cowplot_1.1.1 rjson_0.2.20
#> [58] RColorBrewer_1.1-2 tools_4.0.4 Cairo_1.5-12.2
#> [61] glue_1.4.2 purrr_0.3.4 parallel_4.0.4
#> [64] yaml_2.2.1 clue_0.3-58 colorspace_2.0-0
#> [67] cluster_2.1.1 ComplexHeatmap_2.6.2 knitr_1.31
#> [70] sass_0.3.1