Gene_clustering
library(factoextra)
#> Loading required package: ggplot2
#> Welcome! Want to learn more? See two factoextra-related books at https://goo.gl/ve3WBa
library(COTAN)
library(ggrepel)
library(Rtsne)
library(plotly)
#>
#> Attaching package: 'plotly'
#> The following object is masked from 'package:ggplot2':
#>
#> last_plot
#> The following object is masked from 'package:stats':
#>
#> filter
#> The following object is masked from 'package:graphics':
#>
#> layout
library(tidyverse)
#> ── Attaching packages ─────────────────────────────────────── tidyverse 1.3.1 ──
#> ✓ tibble 3.1.2 ✓ dplyr 1.0.6
#> ✓ tidyr 1.1.3 ✓ stringr 1.4.0
#> ✓ readr 1.4.0 ✓ forcats 0.5.1
#> ✓ purrr 0.3.4
#> ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
#> x dplyr::filter() masks plotly::filter(), stats::filter()
#> x dplyr::lag() masks stats::lag()
library(htmlwidgets)
library(MASS)
#>
#> Attaching package: 'MASS'
#> The following object is masked from 'package:dplyr':
#>
#> select
#> The following object is masked from 'package:plotly':
#>
#> select
library(dendextend)
#>
#> ---------------------
#> Welcome to dendextend version 1.15.1
#> Type citation('dendextend') for how to cite the package.
#>
#> Type browseVignettes(package = 'dendextend') for the package vignette.
#> The github page is: https://github.com/talgalili/dendextend/
#>
#> Suggestions and bug-reports can be submitted at: https://github.com/talgalili/dendextend/issues
#> Or contact: <tal.galili@gmail.com>
#>
#> To suppress this message use: suppressPackageStartupMessages(library(dendextend))
#> ---------------------
#>
#> Attaching package: 'dendextend'
#> The following object is masked from 'package:stats':
#>
#> cutree
library(grid)
library(ggpubr)
#>
#> Attaching package: 'ggpubr'
#> The following object is masked from 'package:dendextend':
#>
#> rotateTo demostrate how to do the gene clustering usign COTAN we begin importing the COTAN object that stores all elaborated data and, in this case, regarding a mouse embrionic cortex dataset (developmental stage E17.5).
input_dir = "Data/"
layers = list("L1"=c("Reln","Lhx5"), "L2/3"=c("Satb2","Cux1"), "L4"=c("Rorb","Sox5") , "L5/6"=c("Bcl11b","Fezf2") , "Prog"=c("Vim","Hes1"))
#objE17 = readRDS(file = paste(input_dir,"E17.5_cortex.cotan.RDS", sep = ""))
objE17 = readRDS(file = paste(input_dir,"E17_cortex_cl2.cotan.RDS", sep = ""))g.space = get.gene.coexpression.space(objE17,
n.genes.for.marker = 25,
primary.markers = unlist(layers))
#> [1] "calculating gene coexpression space: output tanh of reduced coex matrix"
#> L11 L12 L2/31 L2/32 L41 L42 L5/61 L5/62
#> "Reln" "Lhx5" "Satb2" "Cux1" "Rorb" "Sox5" "Bcl11b" "Fezf2"
#> Prog1 Prog2
#> "Vim" "Hes1"
#> [1] "Get p-values on a set of genes on columns genome wide on rows"
#> [1] "Using function S"
#> [1] "function to generate S "
#> [1] "Secondary markers:181"
#> [1] "function to generate S "
#> [1] "Columns (V set) number: 181 Rows (U set) number: 1236"g.space = as.data.frame(as.matrix(g.space))
coex.pca.genes <- prcomp(t(g.space),
center = TRUE,
scale. = F)
fviz_eig(coex.pca.genes, addlabels=TRUE,ncp = 10)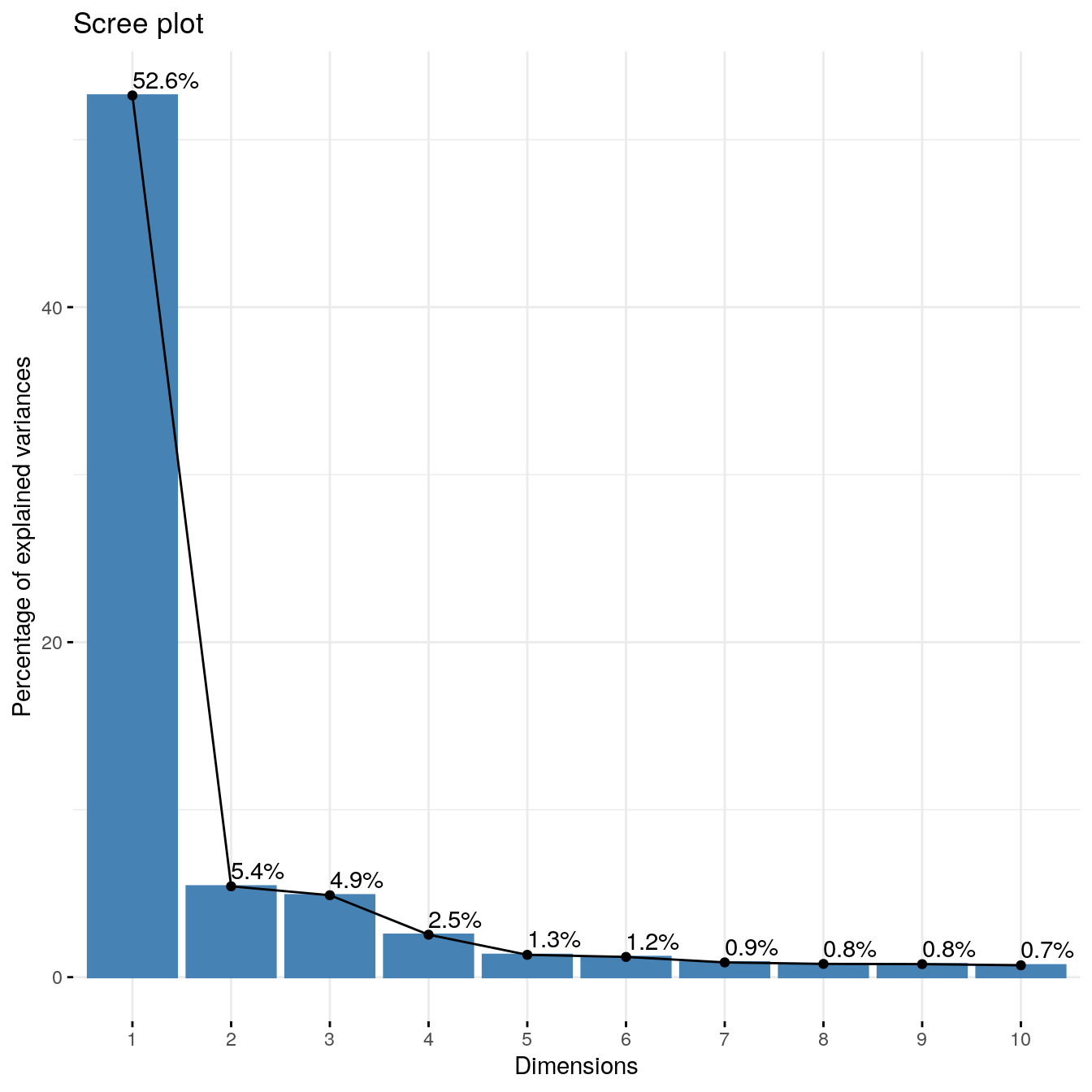
#fviz_eig(coex.pca.genes, choice = "eigenvalue", addlabels=TRUE)Hierarchical clustering
hc.norm = hclust(dist(g.space), method = "ward.D2")
dend <- as.dendrogram(hc.norm)
pca_1 = as.data.frame(coex.pca.genes$rotation[,1:10])
pca_1 = pca_1[order.dendrogram(dend),]
cut = cutree(hc.norm, k = 7, order_clusters_as_data = F)
#- Next lines are only to color and plot the secondary markers
tmp = get.pval(object = objE17,gene.set.col =unlist(layers),gene.set.row = colnames(g.space))
#> L11 L12 L2/31 L2/32 L41 L42 L5/61 L5/62
#> "Reln" "Lhx5" "Satb2" "Cux1" "Rorb" "Sox5" "Bcl11b" "Fezf2"
#> Prog1 Prog2
#> "Vim" "Hes1"
#> [1] "Get p-values on a set of genes on columns on a set of genes on rows"
#> [1] "Using function S"
#> [1] "function to generate S "
for (m in unlist(layers)) {
tmp = as.data.frame(tmp[order(tmp[,m]),])
tmp$rank = c(1:nrow(tmp))
colnames(tmp)[ncol(tmp)] = paste("rank",m,sep = ".")
}
rank.genes = tmp[,(length(unlist(layers))+1):ncol(tmp)]
for (c in c(1:length(colnames(rank.genes)))) {
colnames(rank.genes)[c] =strsplit(colnames(rank.genes)[c], split='.',fixed = T)[[1]][2]
}
L1 = rowSums(rank.genes[,layers[[1]]])
L1[layers[[1]]] = 1
L2 = rowSums(rank.genes[,layers[[2]]])
L2[layers[[2]]] = 1
L4 = rowSums(rank.genes[,layers[[3]]])
L4[layers[[3]]] = 1
L5 =rowSums(rank.genes[,layers[[4]]])
L5[layers[[4]]] = 1
P = rowSums(rank.genes[,layers[[5]]])
P[layers[[5]]] = 1
col.secondary = merge(L1,L2,by="row.names",all.x=TRUE)
colnames(col.secondary)[2:3] = c("L1","L2")
rownames(col.secondary) = col.secondary$Row.names
col.secondary = col.secondary[,2:ncol(col.secondary)]
col.secondary = merge(col.secondary,L4,by="row.names",all.x=TRUE)
colnames(col.secondary)[ncol(col.secondary)] = "L4"
rownames(col.secondary) = col.secondary$Row.names
col.secondary = col.secondary[,2:ncol(col.secondary)]
col.secondary = merge(col.secondary,L5,by="row.names",all.x=TRUE)
colnames(col.secondary)[ncol(col.secondary)] = "L5"
rownames(col.secondary) = col.secondary$Row.names
col.secondary = col.secondary[,2:ncol(col.secondary)]
col.secondary = merge(col.secondary,P,by="row.names",all.x=TRUE)
colnames(col.secondary)[ncol(col.secondary)] = "P"
rownames(col.secondary) = col.secondary$Row.names
col.secondary = col.secondary[,2:ncol(col.secondary)]
# this part is to check that we will color as secondary markers only the genes linked to the
# primary with positive coex
temp.coex = as.matrix(objE17@coex[rownames(objE17@coex) %in% rownames(col.secondary),
colnames(objE17@coex) %in% unlist(layers)])
for (n in rownames(col.secondary)) {
if(any(temp.coex[n,c("Reln","Lhx5")] < 0)){
col.secondary[n,"L1"] = 100000
}
if(any(temp.coex[n,c("Cux1","Satb2")] < 0)){
col.secondary[n,"L2"] = 100000
}
if(any(temp.coex[n,c("Rorb","Sox5")] < 0)){
col.secondary[n,"L4"] = 100000
}
if(any(temp.coex[n,c("Bcl11b","Fezf2")] < 0)){
col.secondary[n,"L5"] = 100000
}
if(any(temp.coex[n,c("Vim","Hes1")] < 0)){
col.secondary[n,"P"] = 100000
}
}
mylist.names <- c("L1", "L2", "L4","L5","P")
pos.link <- vector("list", length(mylist.names))
names(pos.link) <- mylist.names
for (g in rownames(col.secondary)) {
if(length( which(col.secondary[g,] == min(col.secondary[g,]))) == 1 ){
pos.link[[which(col.secondary[g,] == min(col.secondary[g,])) ]] =
c(pos.link[[which(col.secondary[g,] == min(col.secondary[g,])) ]], g)
}
}
# ----
pca_1$highlight = with(pca_1,
ifelse(rownames(pca_1) %in% pos.link$L5, "genes related to L5/6",
ifelse(rownames(pca_1) %in% pos.link$L2 , "genes related to L2/3",
ifelse(rownames(pca_1) %in% pos.link$P , "genes related to Prog" ,
ifelse(rownames(pca_1) %in% pos.link$L1 , "genes related to L1" ,
ifelse(rownames(pca_1) %in% pos.link$L4 ,"genes related to L4" ,
"not marked"))))))
# But sort them based on their order in dend:
#colors_to_use <- pca_1$highlight[order.dendrogram(dend)]
#mycolours <- c("genes related to L5/6" = "#3C5488FF","genes related to L2/3"="#F39B7FFF","genes related to Prog"="#4DBBD5FF","genes related to L1"="#E64B35FF","genes related to L4" = "#91D1C2FF", "not marked"="#B09C85FF")
pca_1$hclust = cut
pca_1$colors = NA
pca_1[pca_1$highlight == "genes related to L5/6", "colors"] = "#3C5488FF"
pca_1[pca_1$highlight == "genes related to L2/3","colors"] = "#F39B7FFF"
pca_1[pca_1$highlight == "genes related to Prog","colors"] = "#4DBBD5FF"
pca_1[pca_1$highlight == "genes related to L1","colors"] = "#E64B35FF"
pca_1[pca_1$highlight == "genes related to L4","colors"] = "#91D1C2FF"
pca_1[pca_1$highlight == "not marked","colors"] = "#B09C85FF"
dend =branches_color(dend,k=7,col=c("#4DBBD5FF","#91D1C2FF","#E64B35FF","gray80","#3C5488FF","#F39B7FFF","gray80" ),groupLabels = T)
dend =color_labels(dend,k=7,labels = rownames(pca_1),col=pca_1$colors)
dend %>%
dendextend::set("labels", ifelse(labels(dend) %in% rownames(pca_1)[rownames(pca_1) %in% colnames(g.space)] ,labels(dend),"")) %>%
# set("branches_k_color", value = c("gray80","#4DBBD5FF","#91D1C2FF" ,"gray80","#F39B7FFF","#E64B35FF","#3C5488FF"), k = 7) %>%
plot(horiz=F, axes=T,ylim = c(0,80))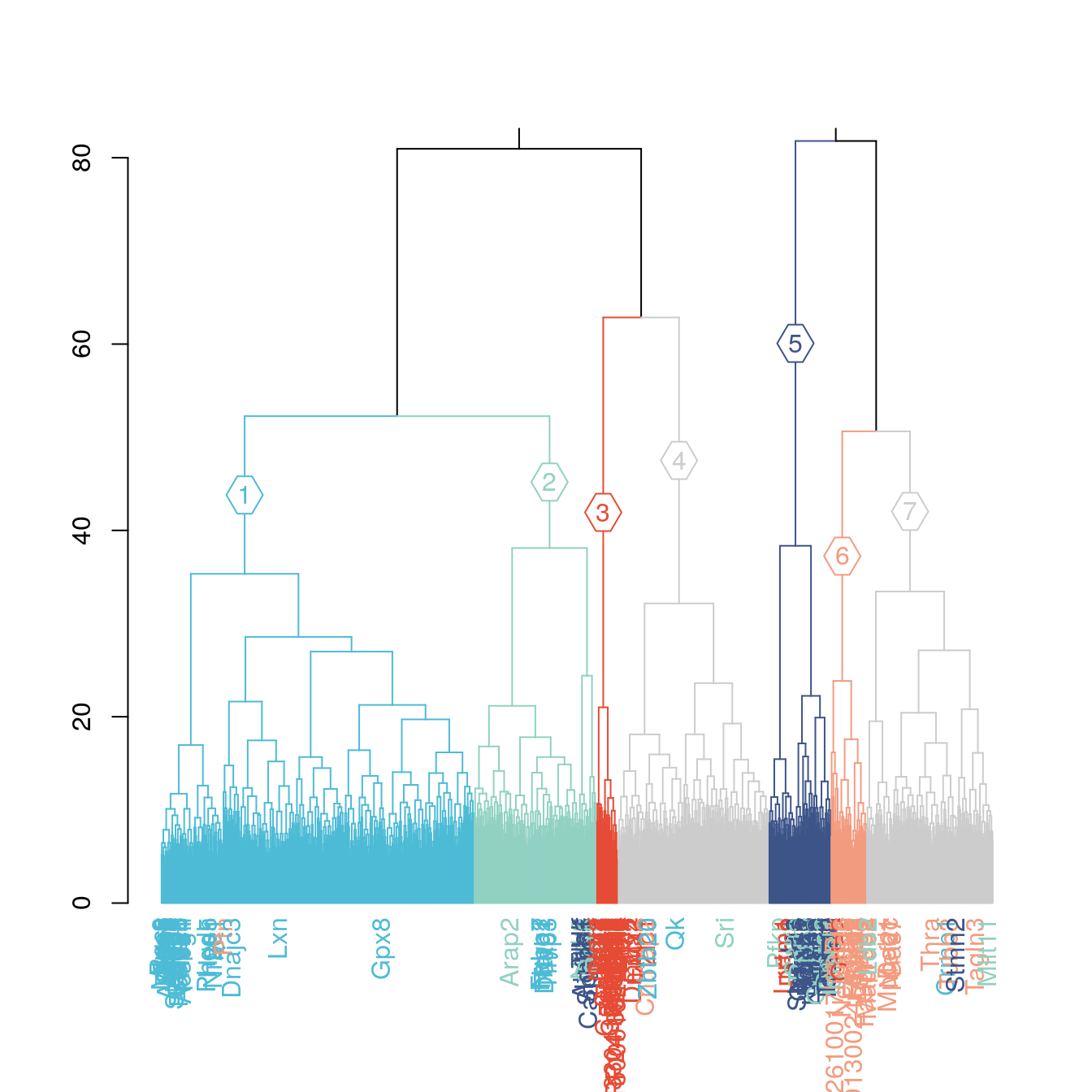
cluster = cut
cluster[cluster == 1] = "#4DBBD5FF"
cluster[cluster == 2] = "#91D1C2FF"
cluster[cluster == 3] = "#E64B35FF"
cluster[cluster == 4] = "#B09C85FF"
cluster[cluster == 5] = "#3C5488FF"
cluster[cluster == 6] = "#F39B7FFF"
cluster[cluster == 7] = "#B09C85FF"
plot.new()
plot(dend,horiz=T, axes=T,xlim = c(100,0),leaflab = "none")
abline(v = 47, lty = 2)
colored_bars(cluster,dend,horiz = T,sort_by_labels_order = F,y_shift = 1,
rowLabels= "" )
gridGraphics::grid.echo()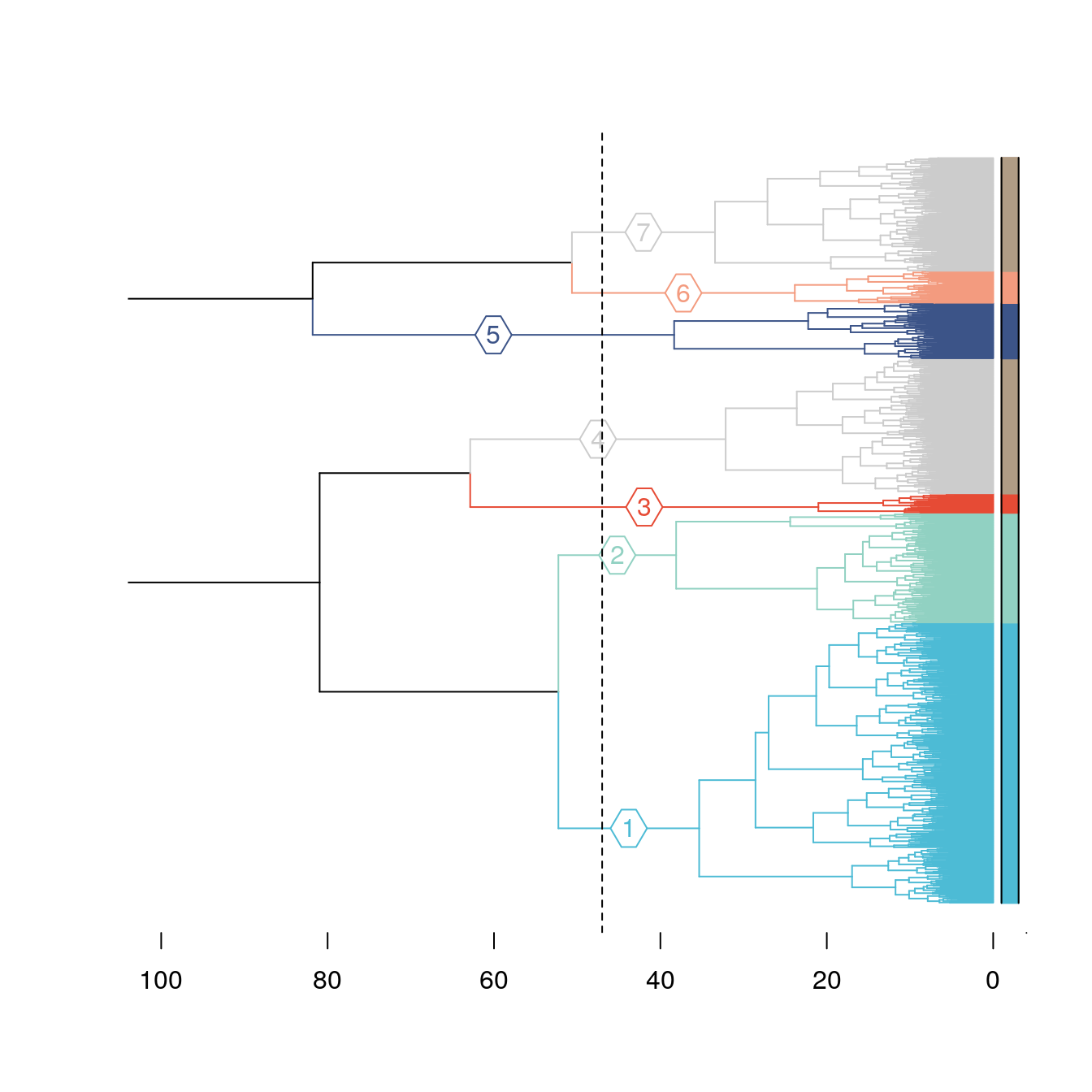
tree <- grid.grab()
par_pca = pca_1[colnames(g.space)[colnames(g.space) %in% rownames(pca_1)],]
#plot N 1
p1 <- ggparagraph(text = paste0(rownames(par_pca[par_pca$hclust == par_pca["Fezf2","hclust"],]), collapse = ", "),
face = "italic",
size =10,
color = "#3C5488FF")
#plot N 2
p2 = ggparagraph(text = paste0(rownames(par_pca[par_pca$hclust == unique(par_pca$hclust)[!unique(par_pca$hclust) %in% unique(par_pca[unlist(layers),"hclust"])][1],]), collapse = ", "),
face = "italic",
size =10,
color = "gray")
#plot N 3
p3 = ggparagraph(text = paste0(rownames(par_pca[par_pca$hclust == unique(par_pca$hclust)[!unique(par_pca$hclust) %in% unique(par_pca[unlist(layers),"hclust"])][2],]), collapse = ", "),
face = "italic",
size =10,
color = "gray")
#plot N 4
p4 = ggparagraph(text = paste0(rownames(par_pca[par_pca$hclust == par_pca["Reln","hclust"],]), collapse = ", "),
face = "italic",
size =10,
color = "#E64B35FF")
#plot N 5
p5 = ggparagraph(text = paste0(rownames(par_pca[par_pca$hclust == par_pca["Cux1","hclust"],]), collapse = ", "),
face = "italic",
size =10,
color = par_pca["Cux1","colors"])
#plot N 6
p6 = ggparagraph(text = paste0(rownames(par_pca[par_pca$hclust == par_pca["Rorb","hclust"],]), collapse = ", "),
face = "italic",
size =10,
color = par_pca["Rorb","colors"])
#plot N 7
p7 = ggparagraph(text = paste0(rownames(par_pca[par_pca$hclust == par_pca["Vim","hclust"],]), collapse = ", "),
face = "italic",
size =10,
color = par_pca["Vim","colors"])
w = ggparagraph(text = " ",
face = "italic",
size =10,
color = "white")
pp =ggarrange(p3,p5,p1,p2,p4,p6,p7,w,
ncol = 1, nrow = 8,
heights = c(0.1,0.15,0.23, 0.1, 0.2, 0.2, 0.1, 0.35))
lay <- rbind(c(1,NA),
c(1,2.5),
c(1,2.5),
c(1,2.5),
c(1,2.5),
c(1,2.5),
c(1,NA))
gridExtra::grid.arrange(tree, pp, layout_matrix = lay)
or just with primary markers
# some more genes as landmarks
controls =list("genes related to L5/6"=c("Foxp2","Tbr1"), "genes related to L2/3"=c("Mef2c"), "genes related to Prog"=c("Nes","Sox2") , "genes related to L1"=c() , "genes related to L4"=c())
dend %>%
dendextend::set("labels", ifelse(labels(dend) %in% rownames(pca_1)[rownames(pca_1) %in% c(unlist(layers),unlist(controls))], labels(dend), "")) %>%
dendextend::set("branches_k_color", value = c("gray80","#4DBBD5FF","#91D1C2FF" ,"gray80","#F39B7FFF","#E64B35FF","#3C5488FF"), k = 7) %>%
plot(horiz=F, axes=T,ylim = c(0,100))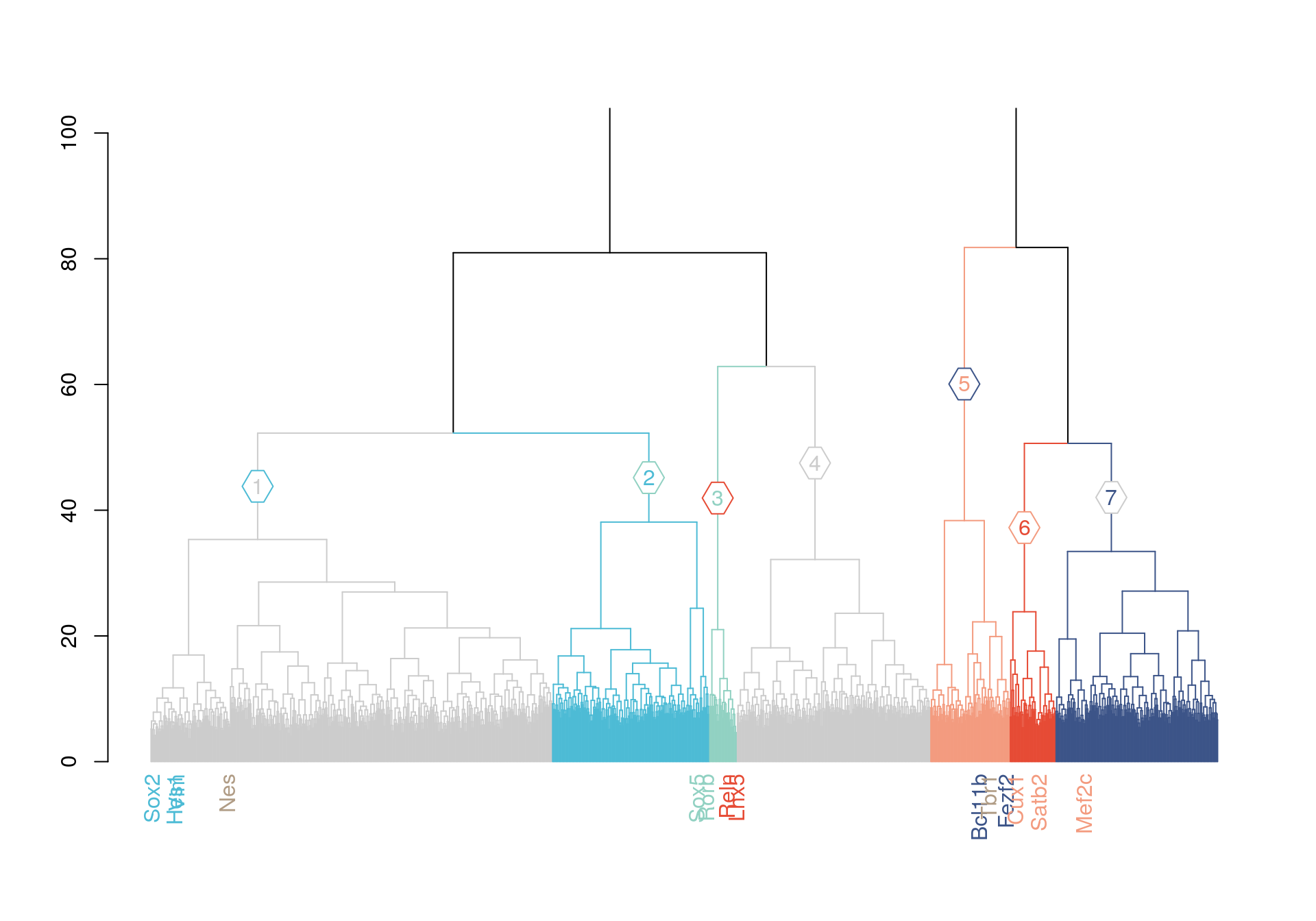
Now we can plot the PCA
# dataframe to be able to label only primary markers and control genes
textdf <- pca_1[rownames(pca_1) %in% c(unlist(layers),unlist(controls)) , ]
for (m in c(1:length(controls))) {
for (g in controls[[m]]) {
if(g %in% rownames(textdf)){
textdf[g,"highlight"] = names(controls[m])
}
}
}
# deciding the colors
mycolours <- c("genes related to L5/6" = "#3C5488FF","genes related to L2/3"="#F39B7FFF","genes related to Prog"="#4DBBD5FF","genes related to L1"="#E64B35FF","genes related to L4" = "#91D1C2FF", "not marked"="#B09C85FF")
# to assing correcly the cluster number and the color
mycolours2 = c("Reln","Satb2","Rorb","Bcl11b","Vim")
names(mycolours2) = unique(cut[unlist(layers)])
mycolours2[mycolours2 == "Reln"] = "#E64B35FF"
mycolours2[mycolours2 == "Satb2"] = "#F39B7FFF"
mycolours2[mycolours2 == "Rorb"] = "#91D1C2FF"
mycolours2[mycolours2 == "Bcl11b"] = "#3C5488FF"
mycolours2[mycolours2 == "Vim"] = "#4DBBD5FF"
color_to_add = unique(pca_1$hclust)[!unique(pca_1$hclust) %in% as.numeric(names(mycolours2))]
names(color_to_add) = unique(pca_1$hclust)[!unique(pca_1$hclust) %in% as.numeric(names(mycolours2))]
color_to_add[color_to_add %in%
unique(pca_1$hclust)[!unique(pca_1$hclust) %in% as.numeric(names(mycolours2))]] = "#B09C85FF"
mycolours2 = c(mycolours2,color_to_add)
pca1 = ggplot(subset(pca_1,!hclust %in% unique(cut[unlist(layers)]) ), aes(x=PC1, y=PC2)) + geom_point(alpha = 0.3,color = "#B09C85FF",size=1)
pca_1$names = rownames(pca_1)
#pca2 = pca1 + geom_point(data = subset(pca_1, highlight != "not marked" ), aes(x=PC1, y=PC2, colour=hclust),size=2.5,alpha = 0.9)
pca2 = pca1 + geom_point(data = subset(pca_1, hclust %in% unique(cut[unlist(layers)]) ), aes(x=PC1, y=PC2, colour=as.character(hclust)),size=1,alpha = 0.5) +
scale_color_manual( "Status", values = mycolours2) +
scale_fill_manual( "Status", values = mycolours2) +
xlab("") + ylab("") +
geom_label_repel(data =textdf , aes(x = PC1, y = PC2, label = rownames(textdf),fill=as.character(hclust)),
label.size = NA,
alpha = 0.5,
direction = "both",
na.rm=TRUE,
seed = 1234) +
geom_label_repel(data =textdf , aes(x = PC1, y = PC2, label = rownames(textdf)),
label.size = NA,
segment.color = 'black',
segment.size = 0.5,
direction = "both",
alpha = 1,
na.rm=TRUE,
fill = NA,
seed = 1234) +
ggtitle("PCA") +
theme_light(base_size=10) +
theme(axis.text.x=element_blank(),plot.title = element_text(size=14,
face="italic",
color="#3C5488FF",
hjust=0.01,
lineheight=1.2,margin = margin(t = 5, b = -15)),
axis.text.y=element_blank(),
legend.position = "none") # titl)
pca2 #+ geom_encircle(data = pca_1, aes(group=hclust)) 
set.seed(NULL)
cell = sample(ncol(objE17@raw),1,replace = T)
print(cell)
#> [1] 461
genes.to.color.red = which(objE17@raw[,cell] > 0)
length(genes.to.color.red)
#> [1] 1199
pca3 = pca2 + geom_point(data = subset(pca_1, names %in% names(genes.to.color.red)), color="red")
pca3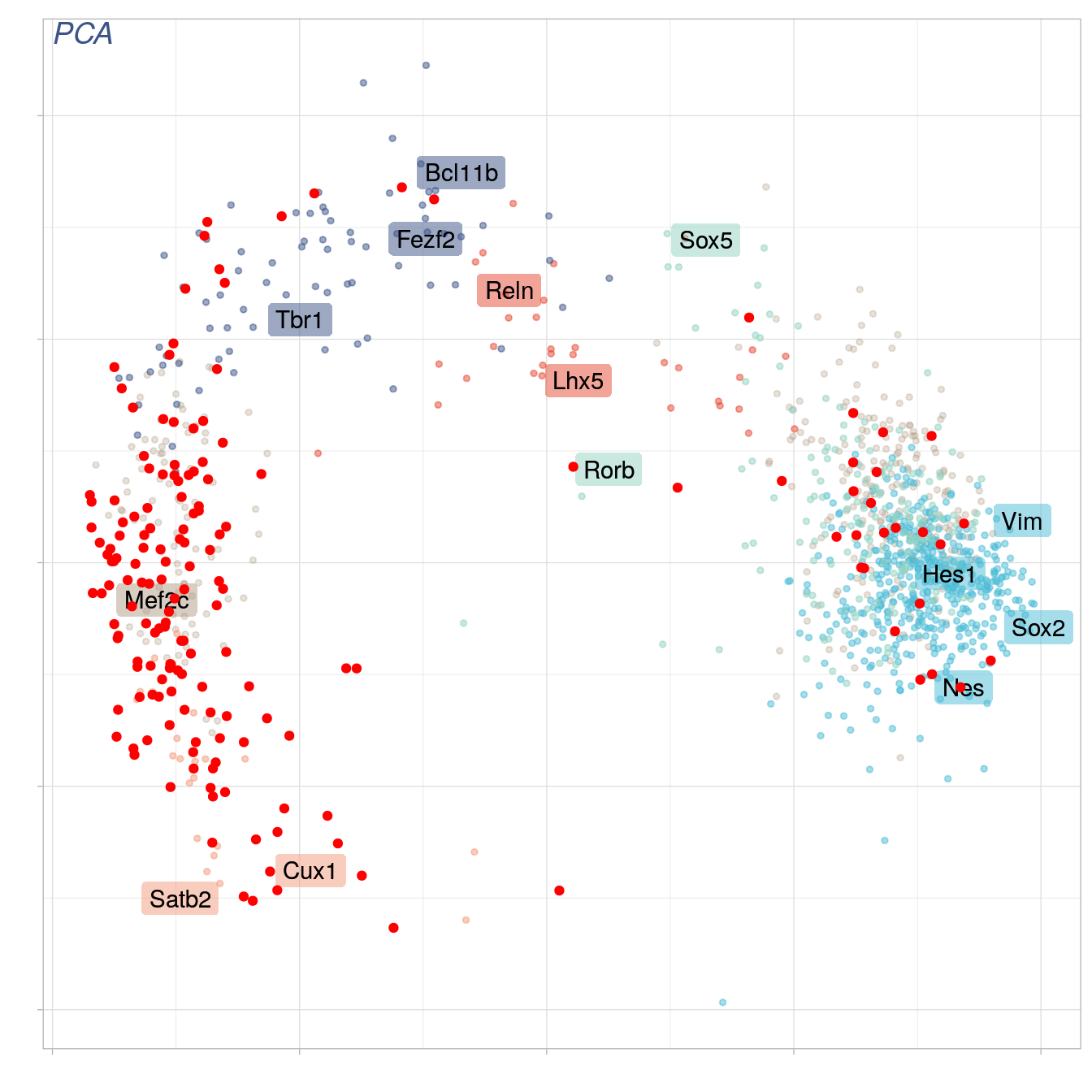
t-SNE code and plot
# run the t-SNE
cl.genes.tsne = Rtsne(g.space ,initial_dims = 100, dims = 2, perplexity=30,eta = 200, verbose=F, max_iter = 3000,theta=0.4,num_threads = 10,pca_center = T, pca_scale = FALSE, normalize = T )
d_tsne_1 = as.data.frame(cl.genes.tsne$Y)
rownames(d_tsne_1) = rownames(g.space)
d_tsne_1 = d_tsne_1[order.dendrogram(dend),]
# save the cluster numebr inside a dataframe with the t-SNE information
d_tsne_1$hclust = cut
d_tsne_1$names = rownames(d_tsne_1)
# as before to label only some genes
textdf <- d_tsne_1[rownames(d_tsne_1) %in% c(unlist(layers),unlist(controls)),]
for (m in c(1:length(controls))) {
for (g in controls[[m]]) {
if(g %in% rownames(textdf)){
textdf[g,"highlight"] = names(controls[m])
}
}
}
p1 = ggplot(subset(d_tsne_1,!hclust %in% unique(cut[unlist(layers)])), aes(x=V1, y=V2)) + geom_point(alpha = 0.3, color = "#B09C85FF", size=1)
p2 = p1 + geom_point(data = subset(d_tsne_1, hclust %in% unique(cut[unlist(layers)]) ), aes(x=V1, y=V2, colour=as.character(hclust)),size=1,alpha = 0.5) +
scale_color_manual("Status", values = mycolours2) +
scale_fill_manual("Status", values = mycolours2) +
xlab("") + ylab("")+
geom_label_repel(data =textdf , aes(x = V1, y = V2, label = names,fill=as.character(hclust)),
label.size = NA,
alpha = 0.5,
direction = "both",
na.rm=TRUE,
seed = 1234) +
geom_label_repel(data =textdf , aes(x = V1, y = V2, label = names),
label.size = NA,
segment.color = 'black',
segment.size = 0.5,
direction = "both",
alpha = 1,
na.rm=TRUE,
fill = NA,
seed = 1234) +
ggtitle("t-SNE") +
theme_light(base_size=10) +
theme(axis.text.x=element_blank(),plot.title = element_text(size=14,
face="italic",
color="#3C5488FF",
hjust=0.01,
lineheight=1.2,margin = margin(t = 5, b = -15)),
axis.text.y=element_blank(),
legend.position = "none") # titl)
p2 Code to create an iteractive plot. This can be modified to be used with all the plots.
Code to create an iteractive plot. This can be modified to be used with all the plots.
p = ggplot(d_tsne_1, aes(x=V1, y=V2, text= paste("gene: ",names))) +
geom_point(size=2, aes(colour=as.character(hclust)), alpha=0.8) +
scale_color_manual("Status", values = mycolours2) +
xlab("") + ylab("") +
ggtitle("t-SNE") +
theme_light(base_size=10) +
theme(axis.text.x=element_blank(),
axis.text.y=element_blank())
ggplotly(p)Multidimensional scaling (MDS) and plot
# run the MDS
genes.dist.euc = dist(g.space, method = "euclidean")
#fit <- isoMDS(genes.dist.euc) # not linear
fit <- isoMDS(genes.dist.euc)
#> initial value 11.623631
#> final value 9.513077
#> converged
fit.genes = as.data.frame(fit$points)
fit.genes = fit.genes[order.dendrogram(dend),]
fit.genes$hclust = cut
fit.genes$names = rownames(fit.genes)
mycolours3 <- c("cluster L5/6 markers" = "#3C5488FF","cluster L2/3 markers"="#F39B7FFF","cluster Prog markers"="#4DBBD5FF","cluster L1 markers"="#E64B35FF","cluster L4 markers" = "#91D1C2FF", "not identified cluster"="#B09C85FF")
#mycolours3 <- c("cluster layer V-VI markers" = "#3C5488FF","cluster layer II-III markers"="#F39B7FFF","cluster progenitor markers"="#4DBBD5FF","cluster layer I markers"="#E64B35FF","cluster layer IV markers" = "#91D1C2FF", "not identified cluster"="#B09C85FF")
#fit.genes$hclust = factor(cutree(hc.norm, 7))
used = vector()
for (k in c(1:length(layers))) {
#print(k)
tt =as.numeric(cut[layers[[k]]][1])
fit.genes[fit.genes$hclust == tt,"cluster"] = paste("cluster",names(layers[k]),"markers", sep = " " )
used = c(used,cut[layers[[k]]][1])
}
fit.genes[fit.genes$hclust %in% (unique(fit.genes$hclust)[!unique(fit.genes$hclust) %in% used]),]$cluster = "not identified cluster"
textdf <- fit.genes[rownames(fit.genes) %in% c(unlist(layers),unlist(controls)),]
f1 = ggplot(subset(fit.genes,!hclust %in% unique(cut[unlist(layers)]) ), aes(x=V1, y=V2)) + geom_point(alpha = 0.3, color = "#B09C85FF", size=1)
f2 = f1 + geom_point(data = subset(fit.genes, hclust %in% unique(cut[unlist(layers)]) ),
aes(x=V1, y=V2, colour=cluster), size=1,alpha = 0.5) +
scale_color_manual("Status", values = mycolours3,
labels = c("Layer I cluster ","Layers II/III cluster",
"Layer IV cluster",
"Layers V/VI cluster",
"Progenitors cluster") ) +
scale_fill_manual("Status", values = mycolours3) +
xlab("") + ylab("")+
geom_label_repel(data =textdf , aes(x = V1, y = V2, label = rownames(textdf),fill=cluster),
label.size = NA,
alpha = 0.5,
direction ="both",
na.rm=TRUE,
seed = 1234, show.legend = FALSE) +
geom_label_repel(data =textdf , aes(x = V1, y = V2, label = rownames(textdf)),
label.size = NA,
segment.color = 'black',
segment.size = 0.5,
direction = "both",
alpha = 1,
na.rm=TRUE,
fill = NA,
seed = 1234, show.legend = FALSE) +
ggtitle("MDS") +
theme_light(base_size=10) +
theme(axis.text.x=element_blank(),plot.title = element_text(size=14,
face="italic",
color="#3C5488FF",
hjust=0.01,
lineheight=1.2,margin = margin(t = 5, b = -15)),
axis.text.y=element_blank(),
legend.title = element_blank(),
legend.text = element_text(color = "#3C5488FF",face ="italic" ),
legend.position = "bottom") # titl)
f2 + scale_y_reverse()+ scale_x_reverse()#+ geom_encircle(data = fit.genes, aes(group=`5_clusters`)) 
sessionInfo()
#> R version 4.1.2 (2021-11-01)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 18.04.6 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/libopenblasp-r0.2.20.so
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] grid stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] ggpubr_0.4.0 dendextend_1.15.1 MASS_7.3-54 htmlwidgets_1.5.3
#> [5] forcats_0.5.1 stringr_1.4.0 dplyr_1.0.6 purrr_0.3.4
#> [9] readr_1.4.0 tidyr_1.1.3 tibble_3.1.2 tidyverse_1.3.1
#> [13] plotly_4.9.3 Rtsne_0.15 ggrepel_0.9.1 COTAN_0.99.7
#> [17] factoextra_1.0.7 ggplot2_3.3.3
#>
#> loaded via a namespace (and not attached):
#> [1] colorspace_2.0-1 ggsignif_0.6.1 rjson_0.2.20
#> [4] ellipsis_0.3.2 rio_0.5.26 circlize_0.4.12
#> [7] GlobalOptions_0.1.2 fs_1.5.0 clue_0.3-59
#> [10] rstudioapi_0.13 farver_2.1.0 fansi_0.4.2
#> [13] lubridate_1.7.10 xml2_1.3.2 codetools_0.2-18
#> [16] doParallel_1.0.16 knitr_1.33 jsonlite_1.7.2
#> [19] Cairo_1.5-12.2 broom_0.7.6 cluster_2.1.2
#> [22] dbplyr_2.1.1 png_0.1-7 compiler_4.1.2
#> [25] httr_1.4.2 basilisk_1.5.0 backports_1.2.1
#> [28] assertthat_0.2.1 Matrix_1.4-0 lazyeval_0.2.2
#> [31] cli_2.5.0 htmltools_0.5.1.1 tools_4.1.2
#> [34] gtable_0.3.0 glue_1.4.2 Rcpp_1.0.6
#> [37] carData_3.0-4 cellranger_1.1.0 jquerylib_0.1.4
#> [40] vctrs_0.3.8 crosstalk_1.1.1 iterators_1.0.13
#> [43] xfun_0.23 openxlsx_4.2.3 rvest_1.0.0
#> [46] lifecycle_1.0.0 rstatix_0.7.0 scales_1.1.1
#> [49] basilisk.utils_1.5.0 hms_1.1.0 parallel_4.1.2
#> [52] RColorBrewer_1.1-2 ComplexHeatmap_2.9.0 yaml_2.2.1
#> [55] curl_4.3.1 reticulate_1.20 gridExtra_2.3
#> [58] stringi_1.6.2 highr_0.9 S4Vectors_0.31.0
#> [61] foreach_1.5.1 BiocGenerics_0.39.0 filelock_1.0.2
#> [64] zip_2.1.1 shape_1.4.6 rlang_0.4.11
#> [67] pkgconfig_2.0.3 matrixStats_0.58.0 evaluate_0.14
#> [70] lattice_0.20-45 labeling_0.4.2 cowplot_1.1.1
#> [73] tidyselect_1.1.1 magrittr_2.0.1 R6_2.5.0
#> [76] IRanges_2.27.0 generics_0.1.0 DBI_1.1.1
#> [79] pillar_1.6.1 haven_2.4.1 foreign_0.8-81
#> [82] withr_2.4.2 abind_1.4-5 dir.expiry_1.1.0
#> [85] modelr_0.1.8 crayon_1.4.1 car_3.0-10
#> [88] utf8_1.2.1 rmarkdown_2.11 viridis_0.6.1
#> [91] GetoptLong_1.0.5 readxl_1.3.1 data.table_1.14.0
#> [94] reprex_2.0.0 digest_0.6.27 gridGraphics_0.5-1
#> [97] stats4_4.1.2 munsell_0.5.0 viridisLite_0.4.0