E15.5 cortex
library(COTAN)
library(data.table)
library(Matrix)
library(ggrepel)
#> Loading required package: ggplot2mycolours <- c(A = "#8491B4B2", B = "#E64B35FF")
my_theme = theme(axis.text.x = element_text(size = 14,
angle = 0, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.text.y = element_text(size = 14,
angle = 0, hjust = 0, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.title.x = element_text(size = 14,
angle = 0, hjust = 0.5, vjust = 0,
face = "plain", colour = "#3C5488FF"),
axis.title.y = element_text(size = 14,
angle = 90, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"))data = as.data.frame(fread(paste(data_dir,
"GSE107122_E155_Combined_Only_Cortical_Cells_DGE.txt.gz",
sep = "/")))
data = as.data.frame(data)
rownames(data) = data$V1
data = data[, 2:ncol(data)]
data[1:10, 1:10]
#> CTGATGGTCTTT CACCGTTAGGAC TAATTCATTCTC TGAGATTATCTT CCCGGCGGCCAC
#> 0610005C13Rik 0 0 0 0 0
#> 0610007N19Rik 0 0 1 0 3
#> 0610007P14Rik 0 1 0 2 1
#> 0610009B22Rik 2 1 0 0 1
#> 0610009D07Rik 4 1 0 3 4
#> 0610009E02Rik 0 0 0 0 0
#> 0610009L18Rik 0 0 0 0 0
#> 0610009O20Rik 1 0 0 0 0
#> 0610010F05Rik 0 5 2 2 0
#> 0610010K14Rik 0 0 0 0 0
#> GTCCTTCCATGT AGCTCGTTTCCA CATATCCATCGC TTACCTTCTCTG ACCTCTCACTTA
#> 0610005C13Rik 0 0 0 0 0
#> 0610007N19Rik 0 0 0 0 0
#> 0610007P14Rik 1 1 3 0 0
#> 0610009B22Rik 0 0 0 0 2
#> 0610009D07Rik 2 3 0 7 2
#> 0610009E02Rik 0 0 0 0 0
#> 0610009L18Rik 1 0 0 0 1
#> 0610009O20Rik 0 0 0 0 0
#> 0610010F05Rik 0 2 0 1 2
#> 0610010K14Rik 0 0 0 0 0Define a directory where the ouput will be stored.
1 Analytical pipeline
Initialize the COTAN object with the row count table and the metadata for the experiment.
obj = new("scCOTAN", raw = data)
obj = initRaw(obj, GEO = "GSE107122", sc.method = "DropSeq",
cond = "mouse cortex E15.5")
#> [1] "Initializing S4 object"Now we can start the cleaning. Analysis requires and starts from a matrix of raw UMI counts after removing possible cell doublets or multiplets and low quality or dying cells (with too high mtRNA percentage, easily done with Seurat or other tools).
If we do not want to consider the mitochondrial genes we can remove them before starting the analysis.
genes_to_rem = rownames(obj@raw[grep("^mt",
rownames(obj@raw)), ]) #genes to remove : mithocondrial
obj@raw = obj@raw[!rownames(obj@raw) %in%
genes_to_rem, ]
cells_to_rem = colnames(obj@raw[which(colSums(obj@raw) ==
0)])
obj@raw = obj@raw[, !colnames(obj@raw) %in%
cells_to_rem]We want also to define a prefix to identify the sample.
t = "E15_cortex_clean"
print(paste("Condition ", t, sep = ""))
#> [1] "Condition E15_cortex_clean"
#--------------------------------------
n_cells = length(colnames(obj@raw))
print(paste("n cells", n_cells, sep = " "))
#> [1] "n cells 2955"
n_it = 1
# obj@raw = obj@raw[rownames(cells),
# colnames(cells)]2 Data cleaning
First, we create the directory to store all information regarding the data cleaning.
if (!file.exists(out_dir)) {
dir.create(file.path(out_dir))
}
if (!file.exists(paste(out_dir, "cleaning",
sep = ""))) {
dir.create(file.path(out_dir, "cleaning"))
}ttm = clean(obj)
#> [1] "Start estimation mu with linear method"
#> [1] 12889 2955
#> rowname AATGCGGACAGG TTCTAAACTTCC TCTTCCCGACGA
#> 5373 Hbb-bs 533.027446 177.779161 274.830198
#> 5372 Hba-a1 142.140652 80.808710 194.782567
#> 5374 Hbb-bt 160.618937 19.394090 24.014289
#> 11759 Tuba1a 5.685626 35.555832 32.019052
#> 11429 Tmsb4x 1.421407 45.252877 13.341272
#> 6514 Malat1 11.371252 22.626439 8.004763
#> 5543 Hnrnpa2b1 5.685626 19.394090 16.009526
#> 9599 Rps5 9.949846 16.161742 8.004763
#> 9517 Rpl32 8.528439 6.464697 16.009526
#> 7448 Nfib 2.842813 19.394090 5.336509
#> 11425 Tmsb10 1.421407 25.858787 0.000000
#> 9487 Rpl13a 9.949846 16.161742 0.000000
#> 11724 Ttc3 2.842813 19.394090 2.668254
#> 7433 Neurod6 0.000000 16.161742 8.004763
#> 5708 Igfbpl1 2.842813 6.464697 13.341272
obj = ttm$object
ttm$pca.cell.2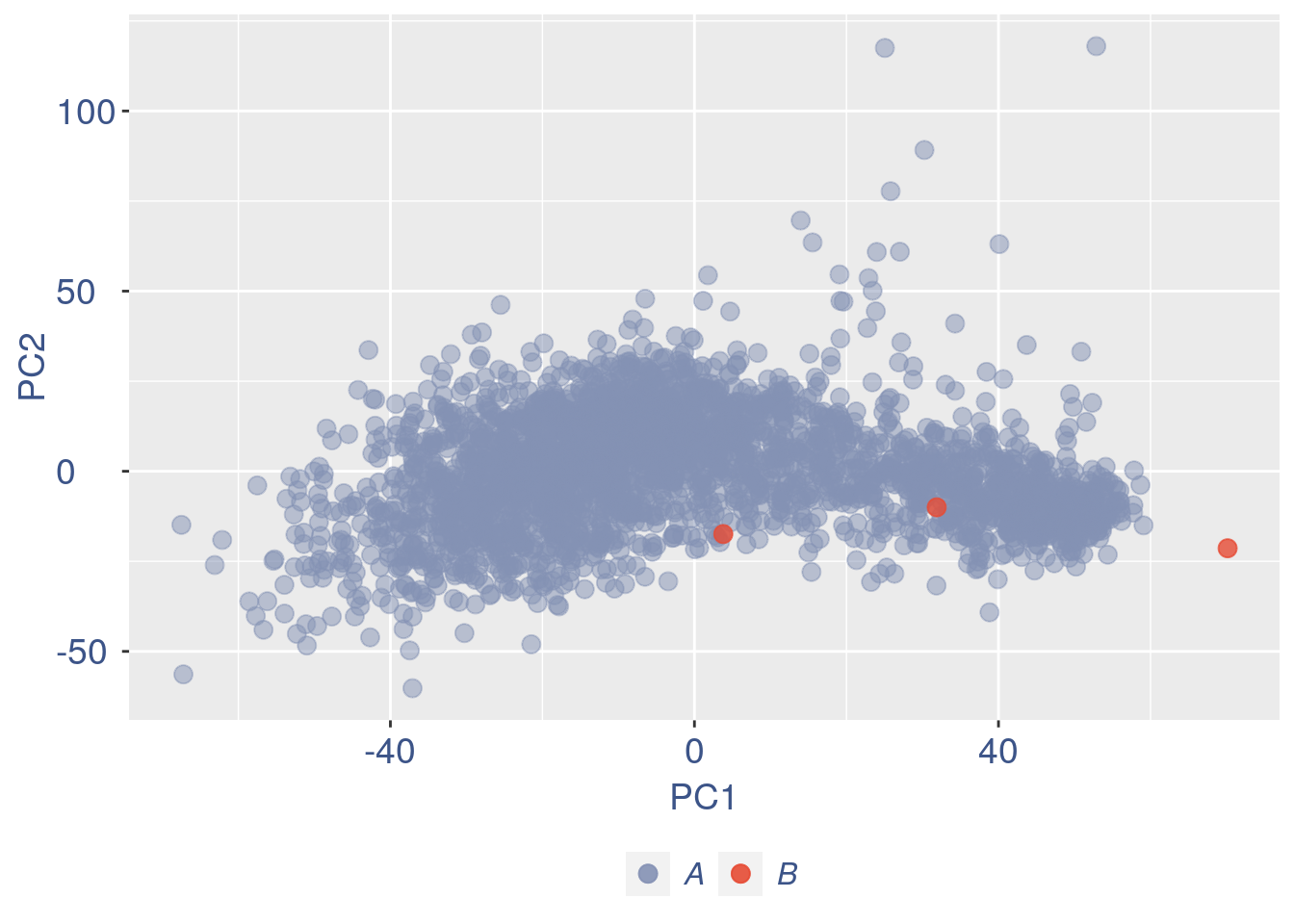
Run this when B cells need to be removed.
pdf(paste(out_dir, "cleaning/", t, "_", n_it,
"_plots_before_cells_exlusion.pdf", sep = ""))
ttm$pca.cell.2
ggplot(ttm$D, aes(x = n, y = means)) + geom_point() +
geom_text_repel(data = subset(ttm$D,
n > (max(ttm$D$n) - 15)), aes(n,
means, label = rownames(ttm$D[ttm$D$n >
(max(ttm$D$n) - 15), ])), nudge_y = 0.05,
nudge_x = 0.05, direction = "x",
angle = 90, vjust = 0, segment.size = 0.2) +
ggtitle("B cell group genes mean expression") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF",
size = 20, face = "italic", vjust = -5,
hjust = 0.02))
dev.off()
#> png
#> 2
if (length(ttm$cl1) < length(ttm$cl2)) {
to_rem = ttm$cl1
} else {
to_rem = ttm$cl2
}
n_it = n_it + 1
obj@raw = obj@raw[, !colnames(obj@raw) %in%
to_rem]
# obj@raw = obj@raw[rownames(obj@raw)
# %in% rownames(cells),colnames(obj@raw)
# %in% colnames(cells)]
gc()
#> used (Mb) gc trigger (Mb) max used (Mb)
#> Ncells 3742647 199.9 6026262 321.9 6026262 321.9
#> Vcells 256234246 1955.0 382282220 2916.6 318501784 2430.0
ttm = clean(obj)
#> [1] "Start estimation mu with linear method"
#> [1] 12887 2952
#> rowname GTGTCGGCGCTN TTACGCACATCT TATTGGCCCATC
#> 6512 Malat1 180.232409 213.042519 234.198856
#> 11757 Tuba1a 28.776603 19.367502 12.774483
#> 10534 Sox4 6.058232 41.501789 12.774483
#> 7532 Nnat 4.543674 41.501789 8.516322
#> 6730 Meg3 1.514558 38.735003 0.000000
#> 7446 Nfib 7.572790 27.667860 4.258161
#> 7431 Neurod6 9.087348 13.833930 12.774483
#> 11165 Tia1 4.543674 0.000000 29.807127
#> 5606 Hsp90ab1 13.631022 5.533572 8.516322
#> 11427 Tmsb4x 9.087348 16.600716 0.000000
#> 7509 Nktr 0.000000 16.600716 8.516322
#> 5225 Gria2 16.660139 2.766786 4.258161
#> 9978 Sfrs18 12.116464 2.766786 8.516322
#> 11047 Tcf4 10.601906 0.000000 12.774483
#> 671 Actb 9.087348 13.833930 0.000000
# ttm = clean.sqrt(obj, cells)
obj = ttm$object
ttm$pca.cell.2
#> png
#> 2
#> used (Mb) gc trigger (Mb) max used (Mb)
#> Ncells 3742887 199.9 6026262 321.9 6026262 321.9
#> Vcells 263308011 2008.9 660874875 5042.1 660872768 5042.1
#> [1] "Start estimation mu with linear method"
#> [1] 12885 2949
#> rowname TATCATGGTGCC GGCGACCTGCCA CATGTCAAAGGC GTTAGGCCCCAC
#> 3762 Fabp7 81.548823 79.2452455 121.248711 80.032947
#> 2916 Dbi 30.166435 18.9971479 30.312178 20.992248
#> 6511 Malat1 8.287482 5.4277565 3.637461 22.304264
#> 11425 Tmsb4x 15.580466 35.2804175 14.549845 7.872093
#> 11755 Tuba1a 15.248967 26.0532314 21.824768 11.808140
#> 8858 Ptn 13.259971 8.6844105 0.000000 44.608528
#> 671 Actb 11.270976 17.9115966 7.274923 6.560078
#> 7089 Mt3 11.270976 11.9410644 20.612281 6.560078
#> 8113 Pea15a 8.950481 5.4277565 9.699897 3.936047
#> 902 Aldoc 4.309491 3.2566539 9.699897 9.184109
#> 2383 Ckb 8.618981 5.4277565 7.274923 6.560078
#> 5605 Hsp90ab1 9.944978 5.4277565 9.699897 6.560078
#> 11751 Ttyh1 5.303989 3.2566539 12.124871 15.744186
#> 1089 Apoe 4.309491 0.5427757 0.000000 39.360466
#> 5540 Hnrnpa2b1 6.298486 6.5133078 2.424974 5.248062
#> ATTTGAGCACGC GCGCCCTTTCCA CGCTAGTTTCGG CCCCATCAGTCG ATCTTTCAGTCG
#> 3762 128.944886 145.410225 79.5134242 79.616006 59.889472
#> 2916 29.756512 30.420549 28.6618157 25.141897 19.963157
#> 6511 16.531396 27.986905 17.5669193 21.789644 73.198244
#> 11425 23.143954 21.902795 14.7931952 20.951580 24.953947
#> 11755 19.837675 20.685974 23.1143675 14.247075 14.972368
#> 8858 23.143954 12.776631 14.7931952 15.923201 13.308772
#> 671 1.653140 14.601864 16.6423446 8.380632 8.317982
#> 7089 6.612558 12.168220 9.2457470 2.514190 3.327193
#> 8113 8.265698 10.342987 10.1703217 11.732885 4.990789
#> 902 11.571977 8.517754 2.7737241 4.190316 11.645175
#> 2383 8.265698 10.951398 6.4720229 6.704506 1.663596
#> 5605 11.571977 4.867288 3.6982988 7.542569 4.990789
#> 11751 9.918837 4.867288 3.6982988 6.704506 0.000000
#> 1089 3.306279 1.825233 0.9245747 2.514190 9.981579
#> 5540 8.265698 9.126165 5.5474482 10.056759 9.981579
#> TTGTGAAGGATC
#> 3762 99.645146
#> 2916 25.185037
#> 6511 7.665011
#> 11425 17.520026
#> 11755 13.140019
#> 8858 9.855014
#> 671 14.235021
#> 7089 7.665011
#> 8113 7.665011
#> 902 7.665011
#> 2383 8.760013
#> 5605 4.380006
#> 11751 6.570010
#> 1089 4.380006
#> 5540 3.285005
Run this only in the last iteration, instead the previous code, when B cells group has not to be removed.
pdf(paste(out_dir, "cleaning/", t, "_", n_it,
"_plots_before_cells_exlusion.pdf", sep = ""))
ttm$pca.cell.2
ggplot(ttm$D, aes(x = n, y = means)) + geom_point() +
geom_text_repel(data = subset(ttm$D,
n > (max(ttm$D$n) - 15)), aes(n,
means, label = rownames(ttm$D[ttm$D$n >
(max(ttm$D$n) - 15), ])), nudge_y = 0.05,
nudge_x = 0.05, direction = "x",
angle = 90, vjust = 0, segment.size = 0.2) +
ggtitle(label = "B cell group genes mean expression",
subtitle = " - B group NOT removed -") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF",
size = 20, face = "italic", vjust = -10,
hjust = 0.02), plot.subtitle = element_text(color = "darkred",
vjust = -15, hjust = 0.01))
dev.off()
#> png
#> 2To colour the pca based on nu_j (so the cells’ efficiency).
nu_est = round(obj@nu, digits = 7)
plot.nu <- ggplot(ttm$pca_cells, aes(x = PC1,
y = PC2, colour = log(nu_est)))
plot.nu = plot.nu + geom_point(size = 1,
alpha = 0.8) + scale_color_gradient2(low = "#E64B35B2",
mid = "#4DBBD5B2", high = "#3C5488B2",
midpoint = log(mean(nu_est)), name = "ln (nu)") +
ggtitle("Cells PCA coloured by cells efficiency") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF",
size = 20), legend.title = element_text(color = "#3C5488FF",
size = 14, face = "italic"), legend.text = element_text(color = "#3C5488FF",
size = 13), legend.key.width = unit(2,
"mm"), legend.position = "right")
pdf(paste(out_dir, "cleaning/", t, "_plots_PCA_efficiency_colored.pdf",
sep = ""))
plot.nu
dev.off()
#> png
#> 2
plot.nu
The next part is use to remove the cells with efficiency too low.
nu_df = data.frame(nu = sort(obj@nu), n = c(1:length(obj@nu)))
ggplot(nu_df, aes(x = n, y = nu)) + geom_point(colour = "#8491B4B2",
size = 1) + my_theme #+ ylim(0,1) + xlim(0,70)
We can zoom on the smallest values and, if we detect a clear elbow, we can decide to remove the cells.
yset = 0.28 #threshold to remove low UDE cells
plot.ude <- ggplot(nu_df, aes(x = n, y = nu)) +
geom_point(colour = "#8491B4B2", size = 1) +
my_theme + ylim(0, 0.5) + xlim(0, 200) +
geom_hline(yintercept = yset, linetype = "dashed",
color = "darkred") + annotate(geom = "text",
x = 500, y = 0.5, label = paste("to remove cells with nu < ",
yset, sep = " "), color = "darkred",
size = 4.5)
pdf(paste(out_dir, "cleaning/", t, "_plots_efficiency.pdf",
sep = ""))
plot.ude
dev.off()
#> png
#> 2
plot.ude
We also save the defined threshold in the metadata and re-run the estimation.
obj@meta[(nrow(obj@meta) + 1), 1:2] = c("Threshold low UDE cells:",
yset)
to_rem = rownames(nu_df[which(nu_df$nu <
yset), ])
obj@raw = obj@raw[, !colnames(obj@raw) %in%
to_rem]Repeat the estimation after the cells are removed.
ttm = clean(obj)
#> [1] "Start estimation mu with linear method"
#> [1] 12882 2936
#> rowname TATCATGGTGCC GGCGACCTGCCA CATGTCAAAGGC GTTAGGCCCCAC
#> 3761 Fabp7 81.820549 79.5092951 121.652718 80.299621
#> 2915 Dbi 30.266951 19.0604475 30.413180 21.062196
#> 6509 Malat1 8.315096 5.4458421 3.649582 22.378583
#> 11423 Tmsb4x 15.632381 35.3979738 14.598326 7.898323
#> 11753 Tuba1a 15.299777 26.1400422 21.897489 11.847485
#> 8856 Ptn 13.304154 8.7133474 0.000000 44.757166
#> 671 Actb 11.308531 17.9712790 7.299163 6.581936
#> 7087 Mt3 11.308531 11.9808527 20.680962 6.581936
#> 8111 Pea15a 8.980304 5.4458421 9.732217 3.949162
#> 902 Aldoc 4.323850 3.2675053 9.732217 9.214711
#> 2383 Ckb 8.647700 5.4458421 7.299163 6.581936
#> 5603 Hsp90ab1 9.978116 5.4458421 9.732217 6.581936
#> 11749 Ttyh1 5.321662 3.2675053 12.165272 15.796647
#> 1089 Apoe 4.323850 0.5445842 0.000000 39.491617
#> 5538 Hnrnpa2b1 6.319473 6.5350106 2.433054 5.265549
#> ATTTGAGCACGC GCGCCCTTTCCA CGCTAGTTTCGG CCCCATCAGTCG ATCTTTCAGTCG
#> 3761 129.374538 145.894741 79.7783674 79.881291 60.089027
#> 2915 29.855663 30.521912 28.7573185 25.225671 20.029676
#> 6509 16.586479 28.080159 17.6254533 21.862248 73.442145
#> 11423 23.221071 21.975777 14.8424870 21.021392 25.037095
#> 11753 19.903775 20.754900 23.1913859 14.294547 15.022257
#> 8856 23.221071 12.819203 14.8424870 15.976258 13.353117
#> 671 1.658648 14.650518 16.6977978 8.408557 8.345698
#> 7087 6.634592 12.208765 9.2765543 2.522567 3.338279
#> 8111 8.293240 10.377450 10.2042098 11.771980 5.007419
#> 902 11.610535 8.546135 2.7829663 4.204278 11.683978
#> 2383 8.293240 10.987888 6.4935880 6.726846 1.669140
#> 5603 11.610535 4.883506 3.7106217 7.567701 5.007419
#> 11749 9.951888 4.883506 3.7106217 6.726846 0.000000
#> 1089 3.317296 1.831315 0.9276554 2.522567 10.014838
#> 5538 8.293240 9.156574 5.5659326 10.090268 10.014838
#> TTGTGAAGGATC
#> 3761 99.977169
#> 2915 25.268955
#> 6509 7.690551
#> 11423 17.578403
#> 11753 13.183803
#> 8856 9.887852
#> 671 14.282453
#> 7087 7.690551
#> 8111 7.690551
#> 902 7.690551
#> 2383 8.789202
#> 5603 4.394601
#> 11749 6.591901
#> 1089 4.394601
#> 5538 3.295951
obj = ttm$object
ttm$pca.cell.2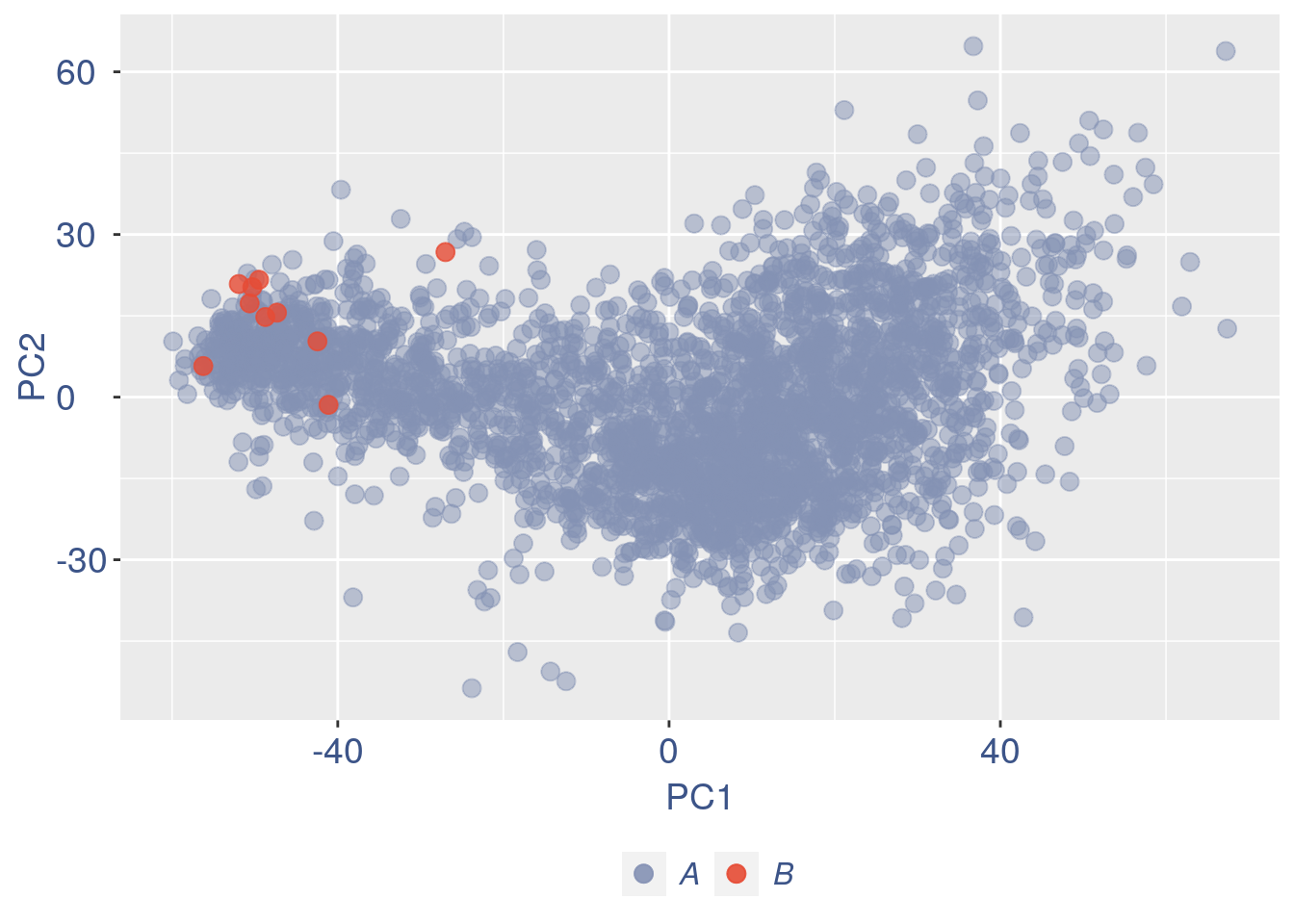
Just to check again, we plot the final efficiency coloured PCA.
nu_est = round(obj@nu, digits = 7)
plot.nu <- ggplot(ttm$pca_cells, aes(x = PC1,
y = PC2, colour = log(nu_est)))
plot.nu = plot.nu + geom_point(size = 2,
alpha = 0.8) + scale_color_gradient2(low = "#E64B35B2",
mid = "#4DBBD5B2", high = "#3C5488B2",
midpoint = log(mean(nu_est)), name = "ln(nu)") +
ggtitle("Cells PCA coloured by cells efficiency: last") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF",
size = 20), legend.title = element_text(color = "#3C5488FF",
size = 14, face = "italic"), legend.text = element_text(color = "#3C5488FF",
size = 13), legend.key.width = unit(2,
"mm"), legend.position = "right")
pdf(paste(out_dir, "cleaning/", t, "_plots_PCA_efficiency_colored_FINAL.pdf",
sep = ""))
plot.nu
dev.off()
#> png
#> 2
plot.nu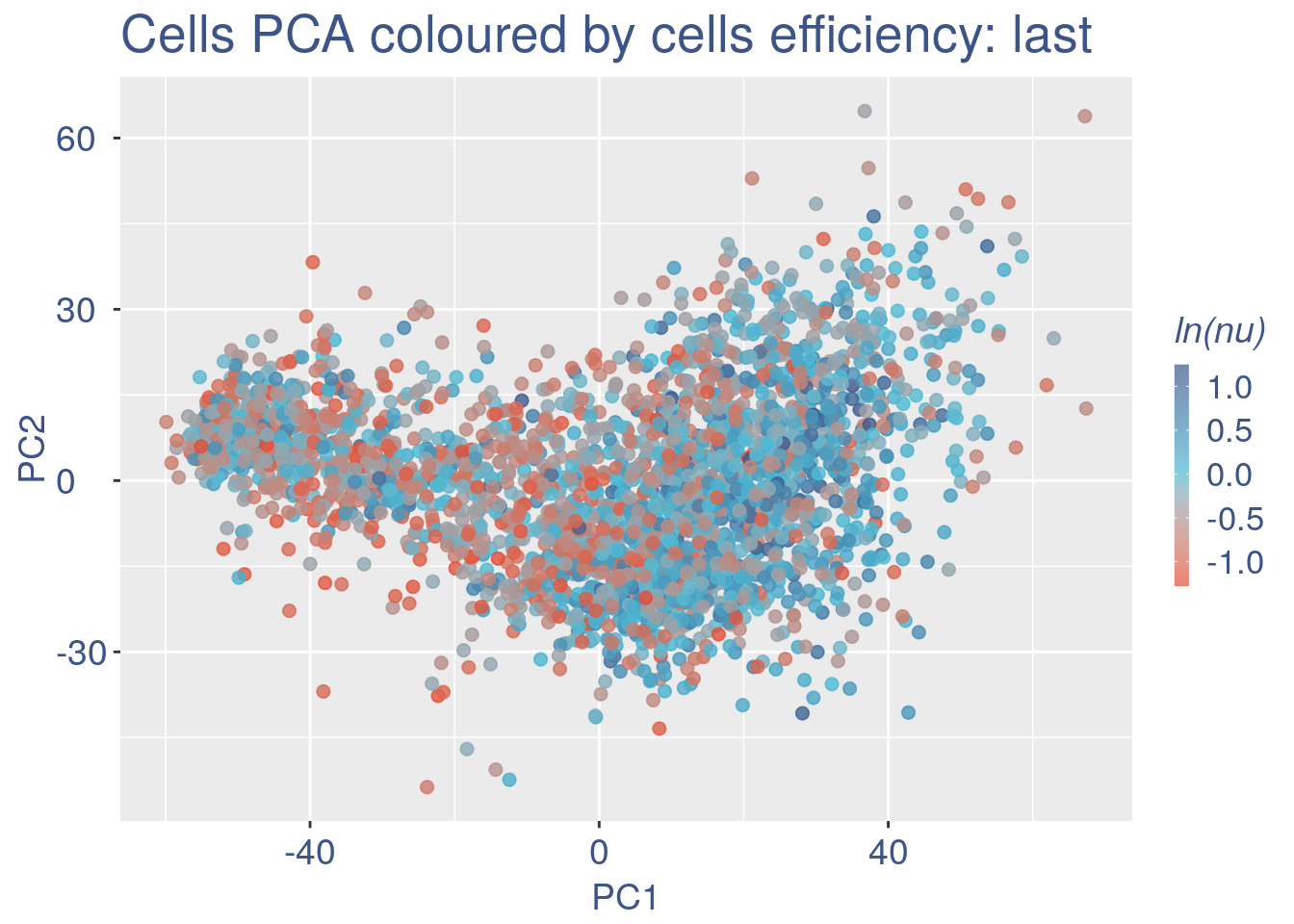
COTAN analysis: in this part all the contingency tables are computed and used to get the statistics (S) To storage efficiency of all the observed tables only the yes_yes is stored. Note that if will be necessary re-computing the yes-yes table, the yes-yes table should be cancelled before running cotan_analysis.
COEX evaluation and storing
saveRDS(obj, file = paste(out_dir, t, ".cotan.RDS",
sep = ""))
obj = get.coex(obj)
# saving the structure
saveRDS(obj, file = paste(out_dir, t, ".cotan.RDS",
sep = ""))Automatic data elaboration (without cleaning)
3 Analysis of the elaborated data
In this case the test possible macroscopic differences between cleaned and not cleaned data.
The initial cell number was:
obj@meta
#> V1 V2
#> 1 GEO: GSE107122
#> 2 scRNAseq method: DropSeq
#> 3 starting n. of cells: 2955
#> 4 Condition sample: mouse cortex E15.5
#> 5 Threshold low UDE cells: 0.28While the final number of cells was:
So were removed:
cells
3.1 GDI with cleaning
quant.p = get.GDI(obj)
#> [1] "function to generate GDI dataframe"
#> [1] "Using S"
#> [1] "function to generate S "
head(quant.p)
#> sum.raw.norm GDI exp.cells
#> 0610007N19Rik 6.029794 2.885667 7.663488
#> 0610007P14Rik 6.541578 1.567536 19.175749
#> 0610009B22Rik 6.522867 1.352988 18.698910
#> 0610009D07Rik 7.938454 1.378622 55.313351
#> 0610009E02Rik 3.924783 1.844979 1.498638
#> 0610009L18Rik 5.167619 1.273583 4.904632In the third column of this data frame is reported the percentage of cells expressing the gene.
NPGs = c("Nes", "Vim", "Sox2", "Sox1", "Notch1",
"Hes1", "Hes5", "Pax6") #,'Hes3'
PNGs = c("Map2", "Tubb3", "Neurod1", "Nefm",
"Nefl", "Dcx", "Tbr1")
hk = c("Calm1", "Cox6b1", "Ppia", "Rpl18",
"Cox7c", "Erh", "H3f3a", "Taf1", "Taf2",
"Gapdh", "Actb", "Golph3", "Mtmr12",
"Zfr", "Sub1", "Tars", "Amacr")
text.size = 12
quant.p$highlight = with(quant.p, ifelse(rownames(quant.p) %in%
NPGs, "NPGs", ifelse(rownames(quant.p) %in%
hk, "Constitutive", ifelse(rownames(quant.p) %in%
PNGs, "PNGs", "normal"))))
textdf <- quant.p[rownames(quant.p) %in%
c(NPGs, hk, PNGs), ]
mycolours <- c(Constitutive = "#00A087FF",
NPGs = "#E64B35FF", PNGs = "#F39B7FFF",
normal = "#8491B4B2")
f1 = ggplot(subset(quant.p, highlight ==
"normal"), aes(x = sum.raw.norm, y = GDI)) +
geom_point(alpha = 0.1, color = "#8491B4B2",
size = 2.5)
GDI_plot = f1 + geom_point(data = subset(quant.p,
highlight != "normal"), aes(x = sum.raw.norm,
y = GDI, colour = highlight), size = 2.5,
alpha = 0.8) + geom_hline(yintercept = quantile(quant.p$GDI)[4],
linetype = "dashed", color = "darkblue") +
geom_hline(yintercept = quantile(quant.p$GDI)[3],
linetype = "dashed", color = "darkblue") +
geom_hline(yintercept = 1.5, linetype = "dotted",
color = "red", size = 0.5) + scale_color_manual("Status",
values = mycolours) + scale_fill_manual("Status",
values = mycolours) + xlab("log normalized counts") +
ylab("GDI") + geom_label_repel(data = textdf,
aes(x = sum.raw.norm, y = GDI, label = rownames(textdf),
fill = highlight), label.size = NA,
alpha = 0.5, direction = "both", na.rm = TRUE,
seed = 1234) + geom_label_repel(data = textdf,
aes(x = sum.raw.norm, y = GDI, label = rownames(textdf)),
label.size = NA, segment.color = "black",
segment.size = 0.5, direction = "both",
alpha = 0.8, na.rm = TRUE, fill = NA,
seed = 1234) + theme(axis.text.x = element_text(size = text.size,
angle = 0, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.text.y = element_text(size = text.size,
angle = 0, hjust = 0, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.title.x = element_text(size = text.size,
angle = 0, hjust = 0.5, vjust = 0,
face = "plain", colour = "#3C5488FF"),
axis.title.y = element_text(size = text.size,
angle = 90, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
legend.title = element_blank(), legend.text = element_text(color = "#3C5488FF",
face = "italic"), legend.position = "bottom") # titl)
legend <- cowplot::get_legend(GDI_plot)
GDI_plot = GDI_plot + theme(legend.position = "none")
GDI_plot
3.2 GDI without cleaning
quant.p = get.GDI(obj2)
#> [1] "function to generate GDI dataframe"
#> [1] "Using S"
#> [1] "function to generate S "
head(quant.p)
#> sum.raw.norm GDI exp.cells
#> 0610007N19Rik 6.025327 2.881709 7.614213
#> 0610007P14Rik 6.537112 1.571894 19.052453
#> 0610009B22Rik 6.524282 1.355656 18.612521
#> 0610009D07Rik 7.935413 1.389342 54.991540
#> 0610009E02Rik 3.920299 1.845393 1.489002
#> 0610009L18Rik 5.163154 1.275495 4.873096In the third column of this data frame is reported the percentage of cells expressing the gene.
NPGs = c("Nes", "Vim", "Sox2", "Sox1", "Notch1",
"Hes1", "Hes5", "Pax6") #,'Hes3'
PNGs = c("Map2", "Tubb3", "Neurod1", "Nefm",
"Nefl", "Dcx", "Tbr1")
hk = c("Calm1", "Cox6b1", "Ppia", "Rpl18",
"Cox7c", "Erh", "H3f3a", "Taf1", "Taf2",
"Gapdh", "Actb", "Golph3", "Mtmr12",
"Zfr", "Sub1", "Tars", "Amacr")
text.size = 12
quant.p$highlight = with(quant.p, ifelse(rownames(quant.p) %in%
NPGs, "NPGs", ifelse(rownames(quant.p) %in%
hk, "Constitutive", ifelse(rownames(quant.p) %in%
PNGs, "PNGs", "normal"))))
textdf <- quant.p[rownames(quant.p) %in%
c(NPGs, hk, PNGs), ]
mycolours <- c(Constitutive = "#00A087FF",
NPGs = "#E64B35FF", PNGs = "#F39B7FFF",
normal = "#8491B4B2")
f1 = ggplot(subset(quant.p, highlight ==
"normal"), aes(x = sum.raw.norm, y = GDI)) +
geom_point(alpha = 0.1, color = "#8491B4B2",
size = 2.5)
GDI_plot = f1 + geom_point(data = subset(quant.p,
highlight != "normal"), aes(x = sum.raw.norm,
y = GDI, colour = highlight), size = 2.5,
alpha = 0.8) + geom_hline(yintercept = quantile(quant.p$GDI)[4],
linetype = "dashed", color = "darkblue") +
geom_hline(yintercept = quantile(quant.p$GDI)[3],
linetype = "dashed", color = "darkblue") +
geom_hline(yintercept = 1.5, linetype = "dotted",
color = "red", size = 0.5) + scale_color_manual("Status",
values = mycolours) + scale_fill_manual("Status",
values = mycolours) + xlab("log normalized counts") +
ylab("GDI") + geom_label_repel(data = textdf,
aes(x = sum.raw.norm, y = GDI, label = rownames(textdf),
fill = highlight), label.size = NA,
alpha = 0.5, direction = "both", na.rm = TRUE,
seed = 1234) + geom_label_repel(data = textdf,
aes(x = sum.raw.norm, y = GDI, label = rownames(textdf)),
label.size = NA, segment.color = "black",
segment.size = 0.5, direction = "both",
alpha = 0.8, na.rm = TRUE, fill = NA,
seed = 1234) + theme(axis.text.x = element_text(size = text.size,
angle = 0, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.text.y = element_text(size = text.size,
angle = 0, hjust = 0, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.title.x = element_text(size = text.size,
angle = 0, hjust = 0.5, vjust = 0,
face = "plain", colour = "#3C5488FF"),
axis.title.y = element_text(size = text.size,
angle = 90, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
legend.title = element_blank(), legend.text = element_text(color = "#3C5488FF",
face = "italic"), legend.position = "bottom") # titl)
legend <- cowplot::get_legend(GDI_plot)
GDI_plot = GDI_plot + theme(legend.position = "none")
GDI_plot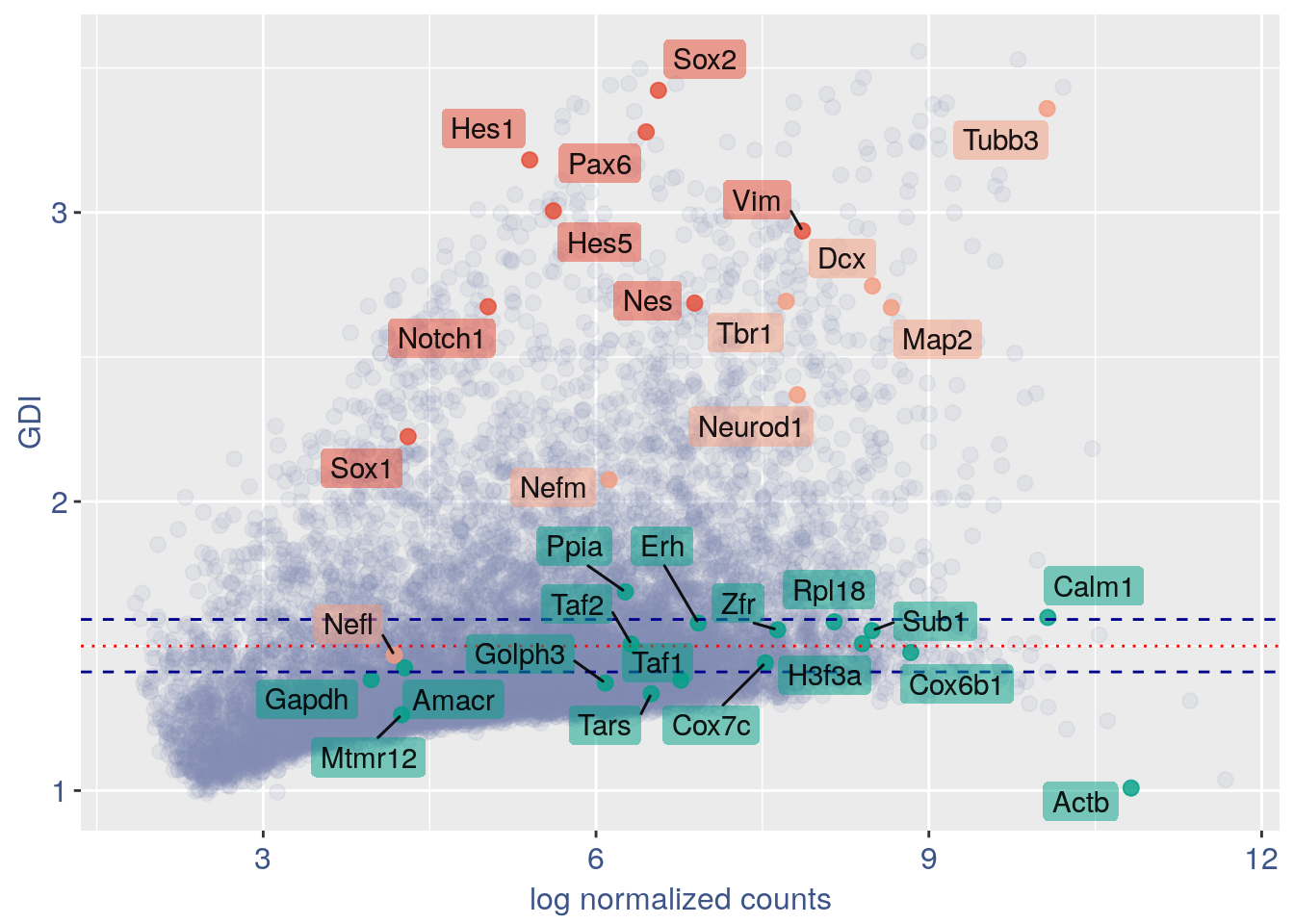
3.3 Heatmaps with cleaning
For the Gene Pair Analysis, we can plot an heatmap with the coex values between two genes sets. To do so we need to define, in a list, the different gene sets (list.genes). Then in the function parameter sets we can decide which sets need to be considered (in the example from 1 to 3). In the condition parameter we should insert an array with each file name prefix to be considered (in the example, the file names is “E15_cortex_clean”).
list.genes = list(Ref.col = PNGs, NPGs = NPGs,
Const. = hk)
plot_heatmap(df_genes = list.genes, sets = c(1:3),
conditions = "E15_cortex_clean", dir = "Data/")
#> [1] "plot heatmap"
#> [1] "Loading condition E15_cortex_clean"
#> [1] "Map2" "Tubb3" "Neurod1" "Nefm" "Nefl" "Dcx" "Tbr1"
#> [1] "Get p-values on a set of genes on columns on a set of genes on rows"
#> [1] "Using function S"
#> [1] "function to generate S "
#> [1] "Ref.col"
#> [1] "NPGs"
#> [1] "Const."
#> [1] "min coex: -0.00726870963092396 max coex 0.00386426580215812"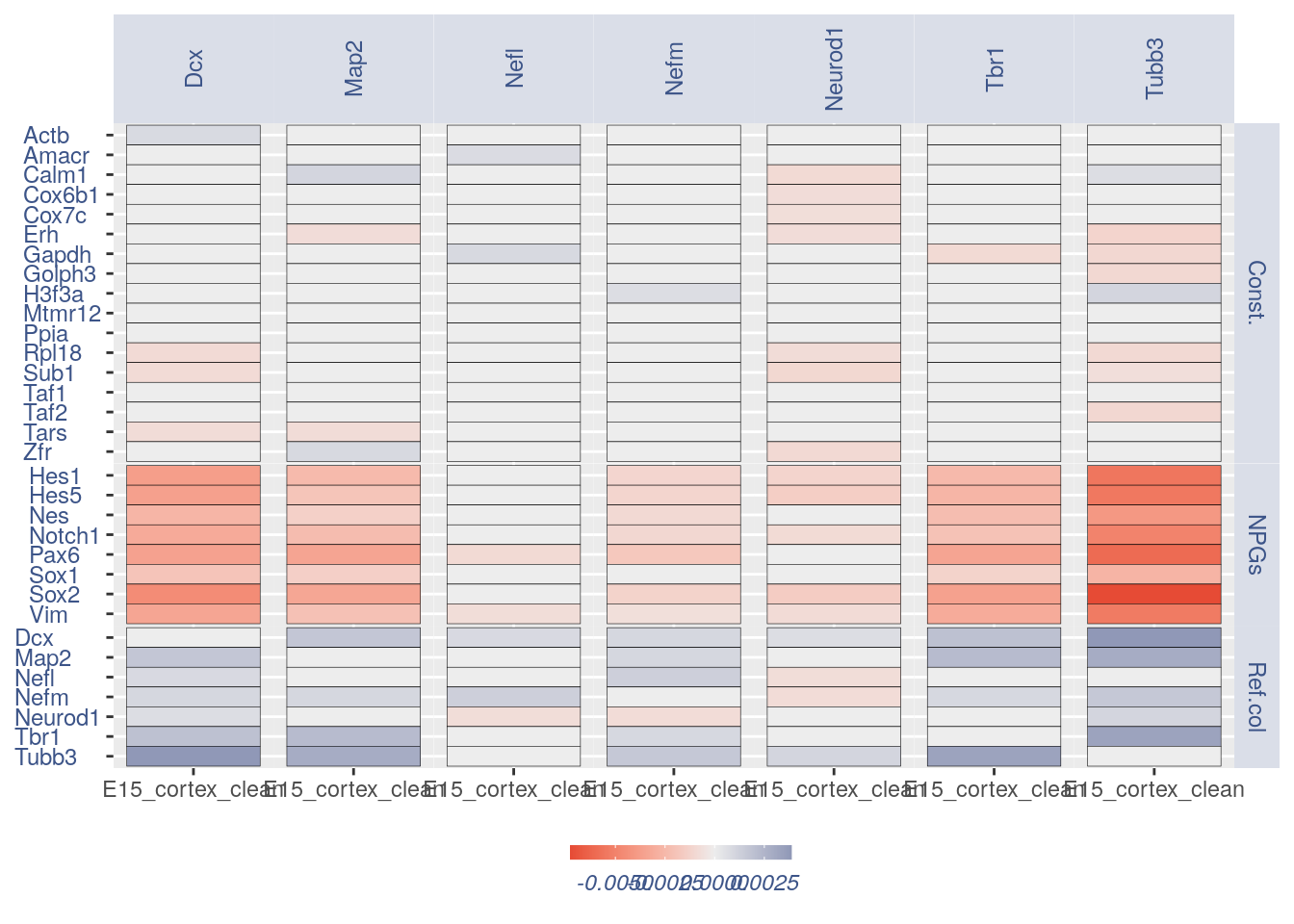
3.4 Heatmaps without cleaning
list.genes = list(Ref.col = PNGs, NPGs = NPGs,
Const. = hk)
plot_heatmap(df_genes = list.genes, sets = c(1:3),
conditions = "E15_cortex", dir = "Data/")
#> [1] "plot heatmap"
#> [1] "Loading condition E15_cortex"
#> [1] "Map2" "Tubb3" "Neurod1" "Nefm" "Nefl" "Dcx" "Tbr1"
#> [1] "Get p-values on a set of genes on columns on a set of genes on rows"
#> [1] "Using function S"
#> [1] "function to generate S "
#> [1] "Ref.col"
#> [1] "NPGs"
#> [1] "Const."
#> [1] "min coex: -0.00724106834694776 max coex 0.00379181054369274"
print(sessionInfo())
#> R version 4.0.4 (2021-02-15)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 18.04.5 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/libopenblasp-r0.2.20.so
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggrepel_0.9.1 ggplot2_3.3.3 Matrix_1.3-2 data.table_1.14.0
#> [5] COTAN_0.1.0
#>
#> loaded via a namespace (and not attached):
#> [1] sass_0.3.1 tidyr_1.1.2 jsonlite_1.7.2
#> [4] R.utils_2.10.1 bslib_0.2.4 assertthat_0.2.1
#> [7] highr_0.8 stats4_4.0.4 yaml_2.2.1
#> [10] pillar_1.5.1 lattice_0.20-41 glue_1.4.2
#> [13] reticulate_1.18 digest_0.6.27 RColorBrewer_1.1-2
#> [16] colorspace_2.0-0 cowplot_1.1.1 htmltools_0.5.1.1
#> [19] R.oo_1.24.0 pkgconfig_2.0.3 GetoptLong_1.0.5
#> [22] purrr_0.3.4 scales_1.1.1 tibble_3.1.0
#> [25] generics_0.1.0 farver_2.1.0 IRanges_2.24.1
#> [28] ellipsis_0.3.1 withr_2.4.1 BiocGenerics_0.36.0
#> [31] magrittr_2.0.1 crayon_1.4.0 evaluate_0.14
#> [34] R.methodsS3_1.8.1 fansi_0.4.2 Cairo_1.5-12.2
#> [37] tools_4.0.4 GlobalOptions_0.1.2 formatR_1.8
#> [40] lifecycle_1.0.0 matrixStats_0.58.0 ComplexHeatmap_2.6.2
#> [43] basilisk.utils_1.2.2 stringr_1.4.0 S4Vectors_0.28.1
#> [46] munsell_0.5.0 cluster_2.1.1 compiler_4.0.4
#> [49] jquerylib_0.1.3 rlang_0.4.10 grid_4.0.4
#> [52] rjson_0.2.20 rappdirs_0.3.3 circlize_0.4.12
#> [55] labeling_0.4.2 rmarkdown_2.7 basilisk_1.2.1
#> [58] gtable_0.3.0 DBI_1.1.1 R6_2.5.0
#> [61] knitr_1.31 dplyr_1.0.4 utf8_1.2.1
#> [64] clue_0.3-58 filelock_1.0.2 shape_1.4.5
#> [67] stringi_1.5.3 parallel_4.0.4 Rcpp_1.0.6
#> [70] vctrs_0.3.6 png_0.1-7 tidyselect_1.1.0
#> [73] xfun_0.22