E13.5 cortex
library(COTAN)
library(data.table)
library(Matrix)
library(ggrepel)
#> Loading required package: ggplot2
# library(latex2exp)mycolours <- c(A = "#8491B4B2", B = "#E64B35FF")
my_theme = theme(axis.text.x = element_text(size = 14,
angle = 0, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.text.y = element_text(size = 14,
angle = 0, hjust = 0, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.title.x = element_text(size = 14,
angle = 0, hjust = 0.5, vjust = 0,
face = "plain", colour = "#3C5488FF"),
axis.title.y = element_text(size = 14,
angle = 90, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"))data = as.data.frame(fread(paste(data_dir,
"GSM2861511_E135_Only_Cortical_Cells_DGE.txt.gz",
sep = "/")))
data = as.data.frame(data)
rownames(data) = data$V1
data = data[, 2:ncol(data)]
data[1:10, 1:10]
#> GTAGCAATTTCT TACTAGATGCTA TATCAGCAGATT TACAGGCCCGTC TGATATACACTT
#> 0610005C13Rik 0 0 0 0 0
#> 0610007N19Rik 0 0 2 0 0
#> 0610007P14Rik 0 0 1 1 0
#> 0610009B22Rik 0 1 0 0 1
#> 0610009D07Rik 0 2 1 0 2
#> 0610009E02Rik 0 0 0 0 0
#> 0610009L18Rik 1 0 0 0 0
#> 0610009O20Rik 0 0 0 0 0
#> 0610010F05Rik 1 4 2 4 1
#> 0610010K14Rik 0 0 0 0 0
#> ATTTCGCGTGAA GGTTTGTCCTTT AACGTCACATCC CCTATCCTTTGC AAATTCGTCGGT
#> 0610005C13Rik 0 0 0 0 0
#> 0610007N19Rik 0 0 0 0 0
#> 0610007P14Rik 1 0 1 0 0
#> 0610009B22Rik 0 0 0 0 0
#> 0610009D07Rik 0 4 2 0 2
#> 0610009E02Rik 0 0 0 0 0
#> 0610009L18Rik 0 0 0 0 0
#> 0610009O20Rik 0 0 0 0 0
#> 0610010F05Rik 2 0 4 3 1
#> 0610010K14Rik 0 0 0 0 0Define a directory where the ouput will be stored.
1 Analytical pipeline
Initialize the COTAN object with the row count table and the metadata for the experiment.
obj = new("scCOTAN", raw = data)
obj = initRaw(obj, GEO = "GSM2861511", sc.method = "DropSeq",
cond = "mouse cortex E13.5")
#> [1] "Initializing S4 object"Now we can start the cleaning. Analysis requires and starts from a matrix of raw UMI counts after removing possible cell doublets or multiplets and low quality or dying cells (with too high mtRNA percentage, easily done with Seurat or other tools).
If we do not want to consider the mitochondrial genes we can remove them before starting the analysis.
genes_to_rem = rownames(obj@raw[grep("^mt",
rownames(obj@raw)), ]) #genes to remove : mithocondrial
obj@raw = obj@raw[!rownames(obj@raw) %in%
genes_to_rem, ]
cells_to_rem = colnames(obj@raw[which(colSums(obj@raw) ==
0)])
obj@raw = obj@raw[, !colnames(obj@raw) %in%
cells_to_rem]We want also to define a prefix to identify the sample.
t = "E13_cortex_clean"
print(paste("Condition ", t, sep = ""))
#> [1] "Condition E13_cortex_clean"
#--------------------------------------
n_cells = length(colnames(obj@raw))
print(paste("n cells", n_cells, sep = " "))
#> [1] "n cells 1137"
n_it = 1
# obj@raw = obj@raw[rownames(cells),
# colnames(cells)]2 Data cleaning
First we create the directory to store all information regardin the data cleaning.
if (!file.exists(out_dir)) {
dir.create(file.path(out_dir))
}
if (!file.exists(paste(out_dir, "cleaning",
sep = ""))) {
dir.create(file.path(out_dir, "cleaning"))
}ttm = clean(obj)
#> [1] "Start estimation mu with linear method"
#> [1] 12725 1137
#> rowname TCTATACAACGA
#> 5286 Hbb-bs 324.117944
#> 5283 Hba-a1 236.415441
#> 5287 Hbb-bt 129.647177
#> 698 Actb 27.963117
#> 11282 Tmsb4x 20.336812
#> 2258 Cfl1 10.168406
#> 5004 Gnas 10.168406
#> 7757 Pabpc1 10.168406
#> 1807 Calm1 8.897355
#> 5611 Igf2 7.626305
#> 6418 Malat1 7.626305
#> 8043 Pfn1 7.626305
#> 9800 Serpinh1 7.626305
#> 10048 Slc25a5 7.626305
#> 10063 Slc2a1 7.626305
obj = ttm$object
ttm$pca.cell.2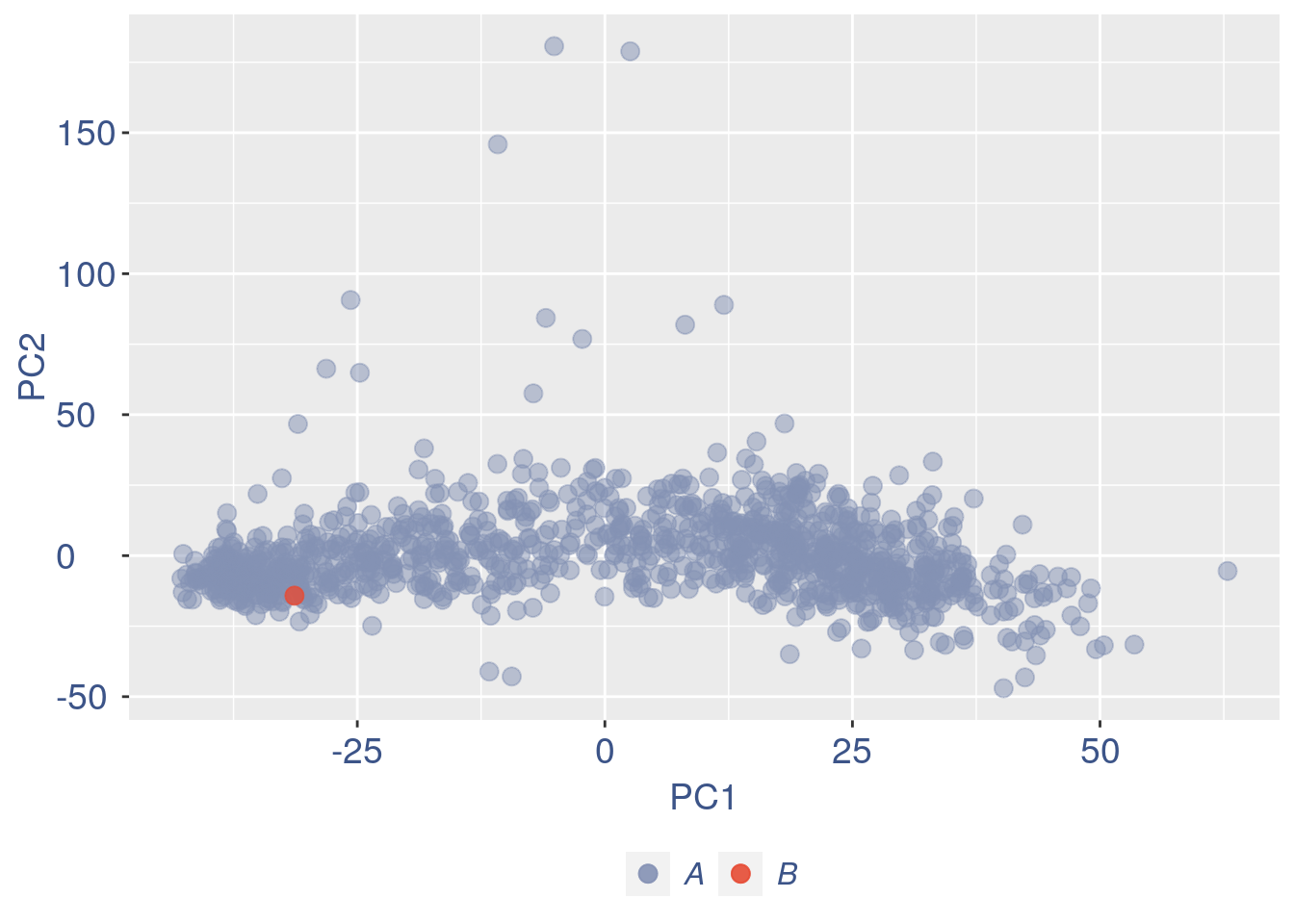
Run this when B cells need to be removed.
pdf(paste(out_dir, "cleaning/", t, "_", n_it,
"_plots_before_cells_exlusion.pdf", sep = ""))
ttm$pca.cell.2
ggplot(ttm$D, aes(x = n, y = means)) + geom_point() +
geom_text_repel(data = subset(ttm$D,
n > (max(ttm$D$n) - 15)), aes(n,
means, label = rownames(ttm$D[ttm$D$n >
(max(ttm$D$n) - 15), ])), nudge_y = 0.05,
nudge_x = 0.05, direction = "x",
angle = 90, vjust = 0, segment.size = 0.2) +
ggtitle("B cell group genes mean expression") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF",
size = 20, face = "italic", vjust = -5,
hjust = 0.02))
dev.off()
#> png
#> 2
if (length(ttm$cl1) < length(ttm$cl2)) {
to_rem = ttm$cl1
} else {
to_rem = ttm$cl2
}
n_it = n_it + 1
obj@raw = obj@raw[, !colnames(obj@raw) %in%
to_rem]
# obj@raw = obj@raw[rownames(obj@raw)
# %in% rownames(cells),colnames(obj@raw)
# %in% colnames(cells)]
gc()
#> used (Mb) gc trigger (Mb) max used (Mb)
#> Ncells 3734347 199.5 6027567 322.0 6027567 322.0
#> Vcells 98496308 751.5 144889565 1105.5 120674318 920.7
ttm = clean(obj)
#> [1] "Start estimation mu with linear method"
#> [1] 12714 1136
#> rowname AAATCCCTAAAG
#> 6410 Malat1 78.03508
#> 7749 Pabpc1 48.77192
#> 3802 Fam168a 39.01754
#> 11592 Ttyh1 39.01754
#> 12214 Zbtb41 39.01754
#> 2720 Ctnna1 29.26315
#> 10789 Taf1d 29.26315
#> 1119 Arglu1 19.50877
#> 1247 Ash1l 19.50877
#> 1717 Bzrap1 19.50877
#> 1789 Cacng4 19.50877
#> 2581 Cox7a2 19.50877
#> 4266 Gas5 19.50877
#> 5075 Gpm6b 19.50877
#> 5606 Igf2bp2 19.50877
# ttm = clean.sqrt(obj, cells)
obj = ttm$object
ttm$pca.cell.2
#> png
#> 2
#> used (Mb) gc trigger (Mb) max used (Mb)
#> Ncells 3734576 199.5 6027567 322.0 6027567 322.0
#> Vcells 100857214 769.5 250660367 1912.4 250659663 1912.4
#> [1] "Start estimation mu with linear method"
#> [1] 12714 1135
#> rowname TTACATTGACCA GAGTTCCTCTTA TTTAAAATACAC
#> 6410 Malat1 164.870988 212.781146 203.06089
#> 6627 Meg3 0.000000 3.940392 43.07352
#> 7334 Nfib 0.000000 0.000000 43.07352
#> 10898 Tcf4 36.065529 0.000000 6.15336
#> 9038 Rbm25 0.000000 11.821175 24.61344
#> 12111 Xist 36.065529 0.000000 0.00000
#> 248 2810055G20Rik 15.456655 15.761566 0.00000
#> 3507 Eif5b 10.304437 19.701958 0.00000
#> 8759 Ptprs 10.304437 0.000000 18.46008
#> 10519 Srrm1 20.608874 7.880783 0.00000
#> 1382 Atrx 10.304437 11.821175 6.15336
#> 11281 Tmx4 0.000000 0.000000 24.61344
#> 5132 Gria2 5.152218 0.000000 18.46008
#> 10522 Srrm4 10.304437 0.000000 12.30672
#> 6038 Kmt2a 0.000000 3.940392 18.46008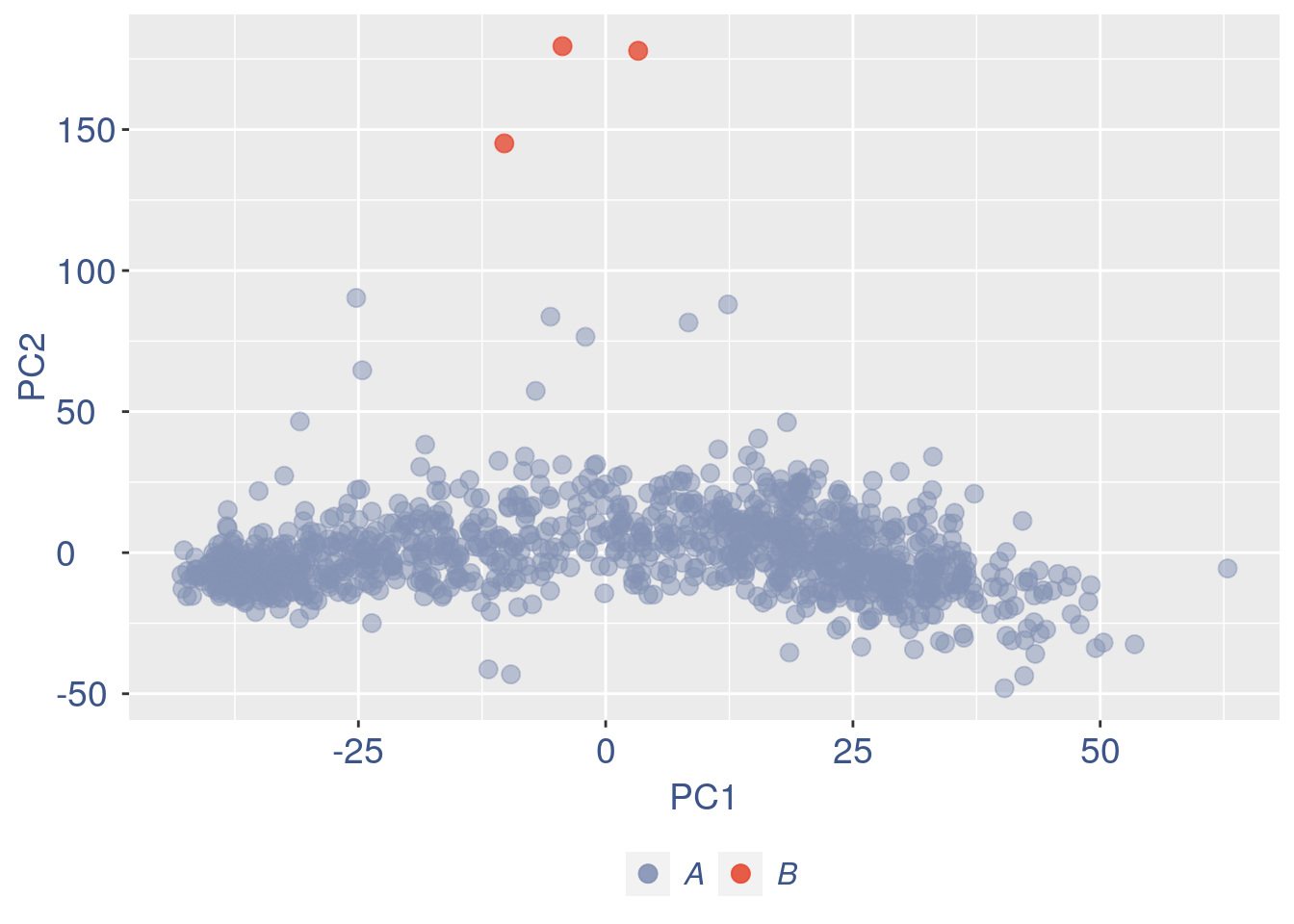
Run this only in the last iteration, instead the previous code, when B cells group has not to be removed.
pdf(paste(out_dir, "cleaning/", t, "_", n_it,
"_plots_before_cells_exlusion.pdf", sep = ""))
ttm$pca.cell.2
ggplot(ttm$D, aes(x = n, y = means)) + geom_point() +
geom_text_repel(data = subset(ttm$D,
n > (max(ttm$D$n) - 15)), aes(n,
means, label = rownames(ttm$D[ttm$D$n >
(max(ttm$D$n) - 15), ])), nudge_y = 0.05,
nudge_x = 0.05, direction = "x",
angle = 90, vjust = 0, segment.size = 0.2) +
ggtitle(label = "B cell group genes mean expression",
subtitle = " - B group NOT removed -") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF",
size = 20, face = "italic", vjust = -10,
hjust = 0.02), plot.subtitle = element_text(color = "darkred",
vjust = -15, hjust = 0.01))
dev.off()
#> png
#> 2To color the pca based on nu_j (so the cells’ efficiency).
nu_est = round(obj@nu, digits = 7)
plot.nu <- ggplot(ttm$pca_cells, aes(x = PC1,
y = PC2, colour = log(nu_est)))
plot.nu = plot.nu + geom_point(size = 1,
alpha = 0.8) + scale_color_gradient2(low = "#E64B35B2",
mid = "#4DBBD5B2", high = "#3C5488B2",
midpoint = log(mean(nu_est)), name = "ln (nu)") +
ggtitle("Cells PCA coloured by cells efficiency") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF",
size = 20), legend.title = element_text(color = "#3C5488FF",
size = 14, face = "italic"), legend.text = element_text(color = "#3C5488FF",
size = 13), legend.key.width = unit(2,
"mm"), legend.position = "right")
pdf(paste(out_dir, "cleaning/", t, "_plots_PCA_efficiency_colored.pdf",
sep = ""))
plot.nu
dev.off()
#> png
#> 2
plot.nu
The next part is use to remove the cells with efficiency too low.
nu_df = data.frame(nu = sort(obj@nu), n = c(1:length(obj@nu)))
ggplot(nu_df, aes(x = n, y = nu)) + geom_point(colour = "#8491B4B2",
size = 1) + my_theme #+ ylim(0,1) + xlim(0,70)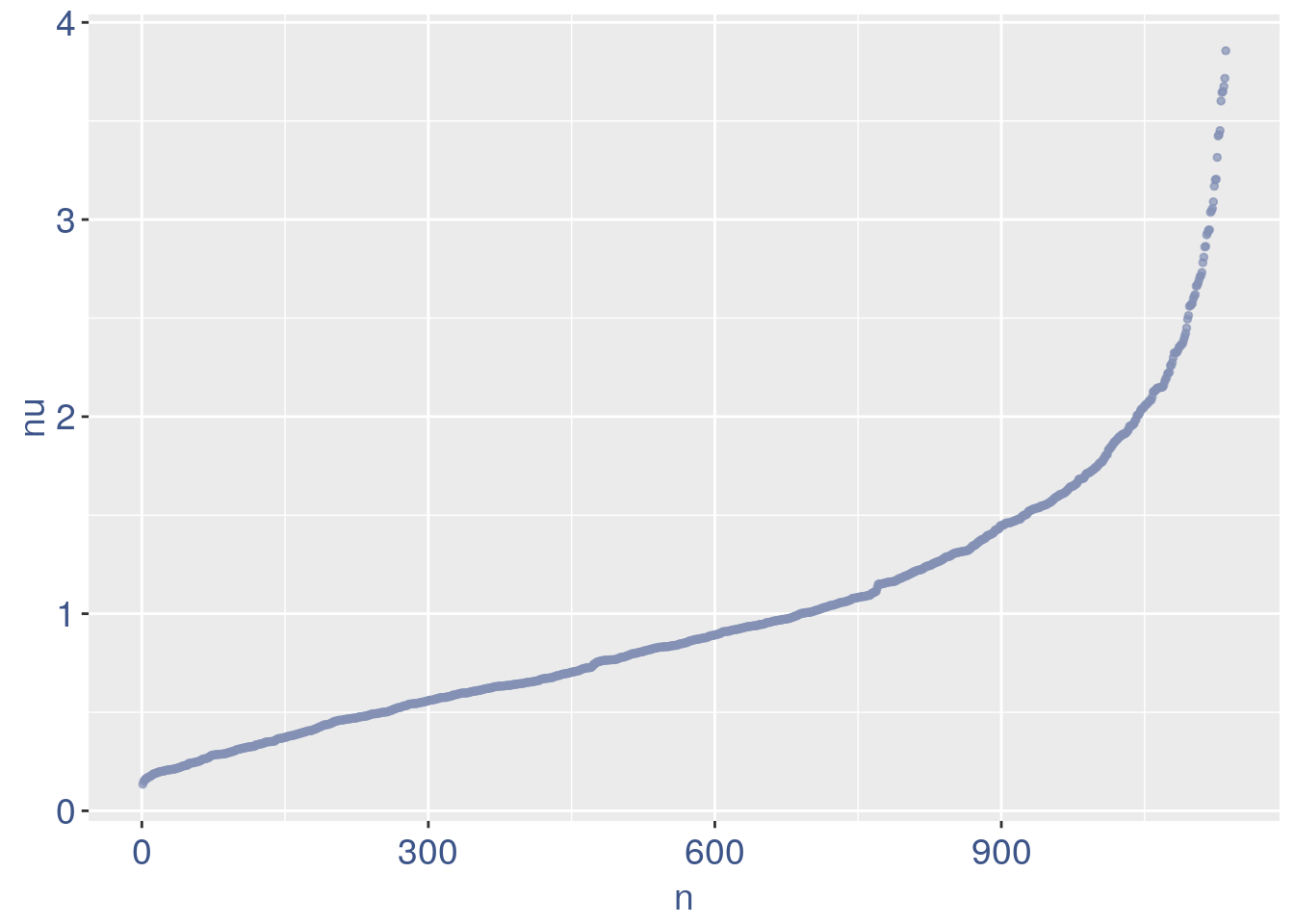
We can zoom on the smalest values and, if we detect a clear elbow, we can decide to remove the cells.
yset = 0.18 #threshold to remove low UDE cells
plot.ude <- ggplot(nu_df, aes(x = n, y = nu)) +
geom_point(colour = "#8491B4B2", size = 1) +
my_theme + ylim(0, 0.5) + xlim(0, 200) +
geom_hline(yintercept = yset, linetype = "dashed",
color = "darkred") + annotate(geom = "text",
x = 500, y = 0.5, label = paste("to remove cells with nu < ",
yset, sep = " "), color = "darkred",
size = 4.5)
pdf(paste(out_dir, "cleaning/", t, "_plots_efficiency.pdf",
sep = ""))
plot.ude
dev.off()
#> png
#> 2
plot.ude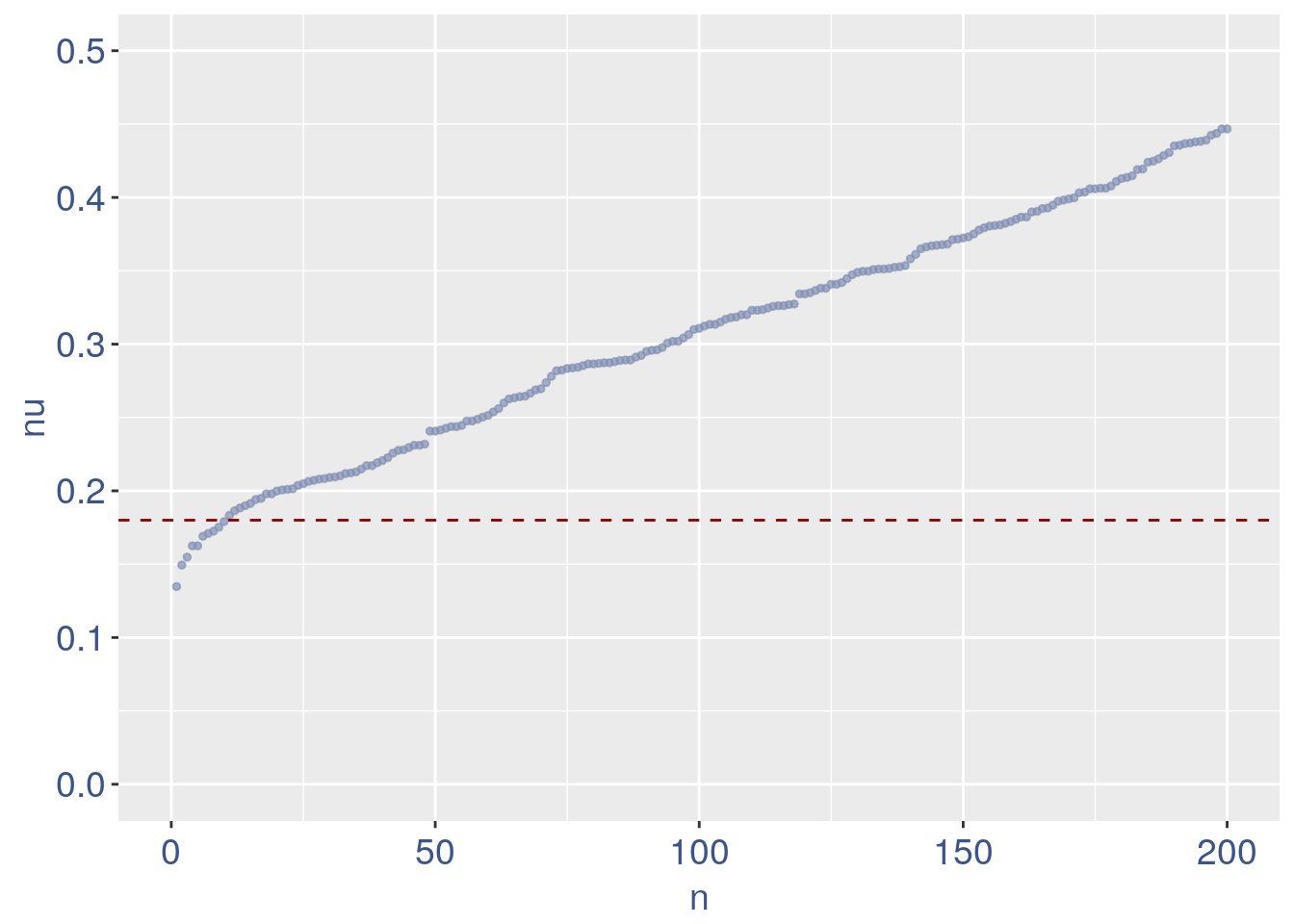
We also save the defined treshold in the metadata and re run the estimation
obj@meta[(nrow(obj@meta) + 1), 1:2] = c("Threshold low UDE cells:",
yset)
to_rem = rownames(nu_df[which(nu_df$nu <
yset), ])
obj@raw = obj@raw[, !colnames(obj@raw) %in%
to_rem]Repeat the estimation after the cells are removed
ttm = clean(obj)
#> [1] "Start estimation mu with linear method"
#> [1] 12708 1125
#> rowname TTACATTGACCA GAGTTCCTCTTA
#> 6405 Malat1 166.09642 214.362683
#> 10892 Tcf4 36.33359 0.000000
#> 12105 Xist 36.33359 0.000000
#> 248 2810055G20Rik 15.57154 15.878717
#> 3504 Eif5b 10.38103 19.848397
#> 10513 Srrm1 20.76205 7.939359
#> 1380 Atrx 10.38103 11.909038
#> 11011 Tial1 10.38103 11.909038
#> 3509 Elavl2 20.76205 0.000000
#> 7414 Nnat 20.76205 0.000000
#> 10203 Smc4 20.76205 0.000000
#> 10369 Sorbs2 20.76205 0.000000
#> 1161 Arid4b 0.00000 19.848397
#> 8594 Prpf38b 15.57154 3.969679
#> 3248 Dpysl3 10.38103 7.939359
obj = ttm$object
ttm$pca.cell.2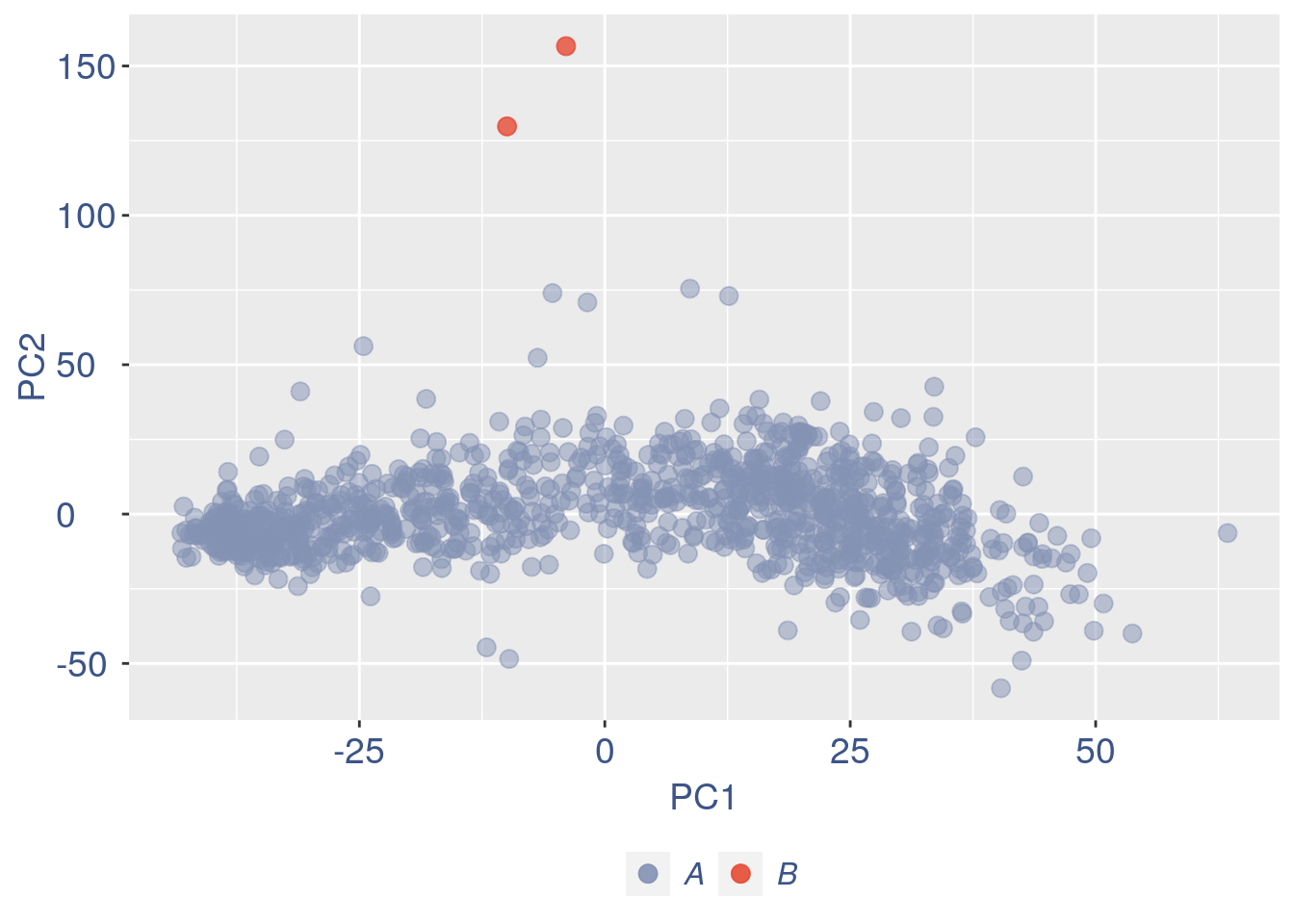
Just to check again, we plot the final efficiency colored PCA
nu_est = round(obj@nu, digits = 7)
plot.nu <- ggplot(ttm$pca_cells, aes(x = PC1,
y = PC2, colour = log(nu_est)))
plot.nu = plot.nu + geom_point(size = 2,
alpha = 0.8) + scale_color_gradient2(low = "#E64B35B2",
mid = "#4DBBD5B2", high = "#3C5488B2",
midpoint = log(mean(nu_est)), name = "ln(nu)") +
ggtitle("Cells PCA coloured by cells efficiency: last") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF",
size = 20), legend.title = element_text(color = "#3C5488FF",
size = 14, face = "italic"), legend.text = element_text(color = "#3C5488FF",
size = 13), legend.key.width = unit(2,
"mm"), legend.position = "right")
pdf(paste(out_dir, "cleaning/", t, "_plots_PCA_efficiency_colored_FINAL.pdf",
sep = ""))
plot.nu
dev.off()
#> png
#> 2
plot.nu
COTAN analysis: in this part all the contingency tables are computed and used to get the statistics (S) To storage efficiency of all the observed tables only the yes_yes is stored. Note that if will be necessary re-computing the yes-yes table, the yes-yes table should be cancelled before running cotan_analysis.
COEX evaluation and storing
saveRDS(obj, file = paste(out_dir, t, ".cotan.RDS",
sep = ""))
obj = get.coex(obj)
# saving the structure
saveRDS(obj, file = paste(out_dir, t, ".cotan.RDS",
sep = ""))Automatic data elaboration (without cleaning)
obj2 = automatic.COTAN.object.creation(df = data,
GEO = "GSM2861510", sc.method = "Drop-seq",
cond = "E13_cortex", out_dir = "Data/")
#> [1] "Initializing S4 object"
#> [1] "Condition E13_cortex"
#> [1] "n cells 1137"
#> [1] "Start estimation mu with linear method"
#> [1] 12725 1137
#> rowname TCTATACAACGA
#> 5286 Hbb-bs 324.117944
#> 5283 Hba-a1 236.415441
#> 5287 Hbb-bt 129.647177
#> 698 Actb 27.963117
#> 11282 Tmsb4x 20.336812
#> 2258 Cfl1 10.168406
#> 5004 Gnas 10.168406
#> 7757 Pabpc1 10.168406
#> 1807 Calm1 8.897355
#> 5611 Igf2 7.626305
#> 6418 Malat1 7.626305
#> 8043 Pfn1 7.626305
#> 9800 Serpinh1 7.626305
#> 10048 Slc25a5 7.626305
#> 10063 Slc2a1 7.626305
#> [1] "Cotan analysis function started"
#> [1] "cotan analysis"
#> [1] "mu estimator creation"
#> [1] "save effective constitutive genes"
#> [1] "start a minimization"
#> [1] "Next gene: Calml3 number 1810"
#> [1] "Next gene: Fam181a number 3820"
#> [1] "Next gene: Kank2 number 5830"
#> [1] "Next gene: Parva number 7840"
#> [1] "Next gene: Sfxn1 number 9850"
#> [1] "Next gene: Usp31 number 11860"
#> [1] "Final trance!"
#> [1] "a min: -0.0412673950195312 | a max 748.25 | negative a %: 10.9705304518664"
#> [1] "Only analysis time 0.786702223618825"
#> [1] "Cotan coex estimation started"
#> [1] "coex dataframe creation"
#> [1] "Generating contingency tables for observed data"
#> [1] "creation of observed yes/yes contingency table"
#> [1] "mu estimator creation"
#> [1] "expected contingency tables creation"
#> [1] "The distance between estimated n of zeros and observed number of zero is 0.0602744425111541 over 12725"
#> [1] "Done"
#> [1] "coex estimation"
#> [1] "Diagonal coex values substituted with 0"
#> [1] "Total time 2.61206785837809"
#> [1] "Only coex time 1.70640178918838"
#> [1] "Saving elaborated data locally at Data/E13_cortex.cotan.RDS"3 Analysis of the elaborated data
In this case the test possible macroscopic differences between cleaned and not cleaned data.
The initial cell number was:
obj = readRDS(paste(out_dir, t, ".cotan.RDS",
sep = ""))
obj@meta
#> V1 V2
#> 1 GEO: GSM2861511
#> 2 scRNAseq method: DropSeq
#> 3 starting n. of cells: 1137
#> 4 Condition sample: mouse cortex E13.5
#> 5 Threshold low UDE cells: 0.18While the final number of cells was:
So were removed:
cells
3.1 GDI with cleaning
quant.p = get.GDI(obj)
#> [1] "function to generate GDI dataframe"
#> [1] "Using S"
#> [1] "function to generate S "
head(quant.p)
#> sum.raw.norm GDI exp.cells
#> 0610007N19Rik 5.166481 2.188999 10.133333
#> 0610007P14Rik 5.362836 1.677978 16.266667
#> 0610009B22Rik 5.375005 1.298590 15.022222
#> 0610009D07Rik 6.813781 1.414547 46.044444
#> 0610009E02Rik 3.686037 1.789690 3.111111
#> 0610009L18Rik 3.269327 1.231394 2.400000In the third column of this data frame is reported the percentage of cells expressing the gene.
NPGs = c("Nes", "Vim", "Sox2", "Sox1", "Notch1",
"Hes1", "Hes5", "Pax6") #,'Hes3'
PNGs = c("Map2", "Tubb3", "Neurod1", "Nefm",
"Nefl", "Dcx", "Tbr1")
hk = c("Calm1", "Cox6b1", "Ppia", "Rpl18",
"Cox7c", "Erh", "H3f3a", "Taf1", "Taf2",
"Gapdh", "Actb", "Golph3", "Mtmr12",
"Zfr", "Sub1", "Tars", "Amacr")
text.size = 12
quant.p$highlight = with(quant.p, ifelse(rownames(quant.p) %in%
NPGs, "NPGs", ifelse(rownames(quant.p) %in%
hk, "Constitutive", ifelse(rownames(quant.p) %in%
PNGs, "PNGs", "normal"))))
textdf <- quant.p[rownames(quant.p) %in%
c(NPGs, hk, PNGs), ]
mycolours <- c(Constitutive = "#00A087FF",
NPGs = "#E64B35FF", PNGs = "#F39B7FFF",
normal = "#8491B4B2")
f1 = ggplot(subset(quant.p, highlight ==
"normal"), aes(x = sum.raw.norm, y = GDI)) +
geom_point(alpha = 0.1, color = "#8491B4B2",
size = 2.5)
GDI_plot = f1 + geom_point(data = subset(quant.p,
highlight != "normal"), aes(x = sum.raw.norm,
y = GDI, colour = highlight), size = 2.5,
alpha = 0.8) + geom_hline(yintercept = quantile(quant.p$GDI)[4],
linetype = "dashed", color = "darkblue") +
geom_hline(yintercept = quantile(quant.p$GDI)[3],
linetype = "dashed", color = "darkblue") +
geom_hline(yintercept = 1.5, linetype = "dotted",
color = "red", size = 0.5) + scale_color_manual("Status",
values = mycolours) + scale_fill_manual("Status",
values = mycolours) + xlab("log normalized counts") +
ylab("GDI") + geom_label_repel(data = textdf,
aes(x = sum.raw.norm, y = GDI, label = rownames(textdf),
fill = highlight), label.size = NA,
alpha = 0.5, direction = "both", na.rm = TRUE,
seed = 1234) + geom_label_repel(data = textdf,
aes(x = sum.raw.norm, y = GDI, label = rownames(textdf)),
label.size = NA, segment.color = "black",
segment.size = 0.5, direction = "both",
alpha = 0.8, na.rm = TRUE, fill = NA,
seed = 1234) + theme(axis.text.x = element_text(size = text.size,
angle = 0, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.text.y = element_text(size = text.size,
angle = 0, hjust = 0, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.title.x = element_text(size = text.size,
angle = 0, hjust = 0.5, vjust = 0,
face = "plain", colour = "#3C5488FF"),
axis.title.y = element_text(size = text.size,
angle = 90, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
legend.title = element_blank(), legend.text = element_text(color = "#3C5488FF",
face = "italic"), legend.position = "bottom") # titl)
legend <- cowplot::get_legend(GDI_plot)
GDI_plot = GDI_plot + theme(legend.position = "none")
GDI_plot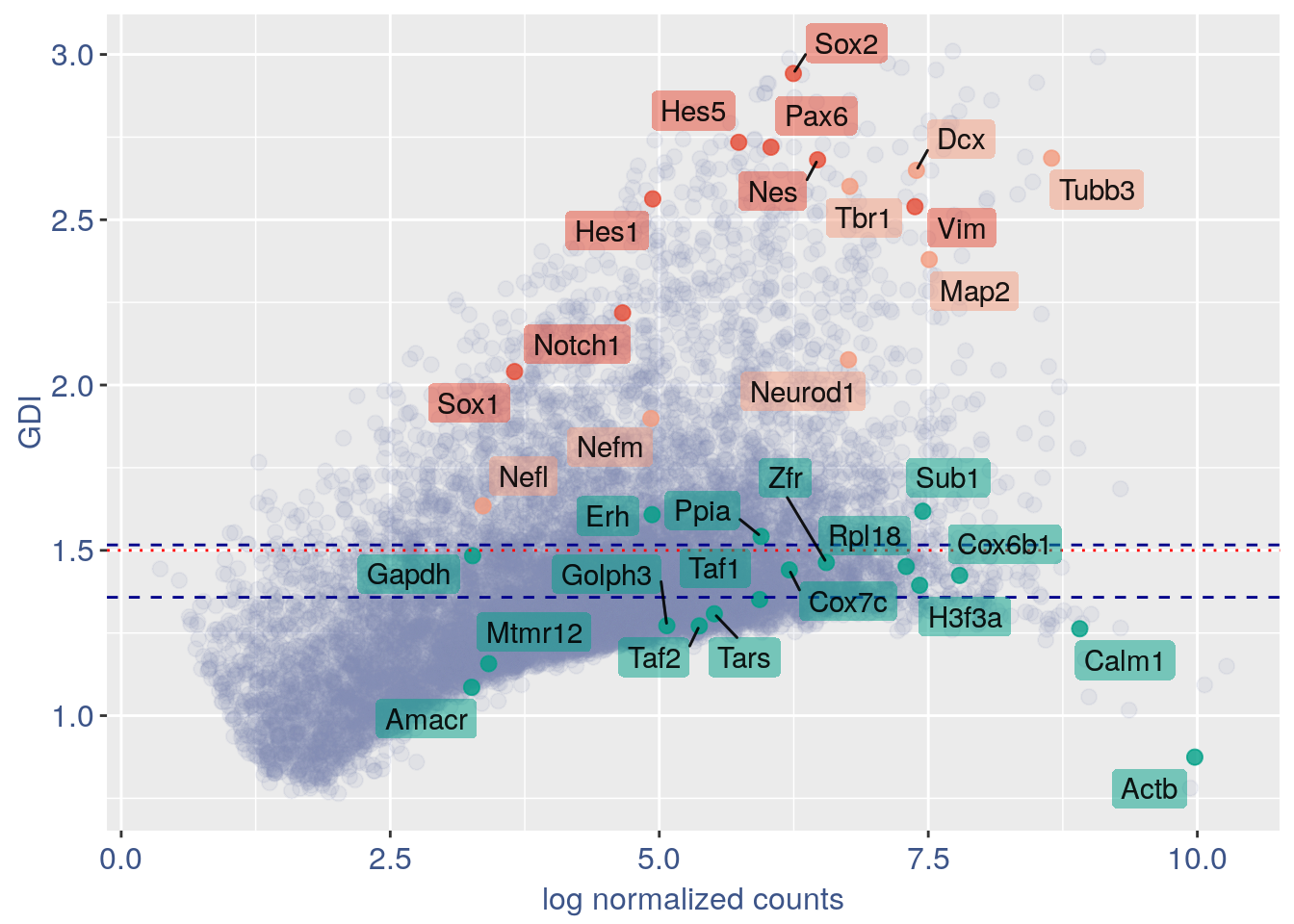
3.2 GDI without cleaning
quant.p = get.GDI(obj2)
#> [1] "function to generate GDI dataframe"
#> [1] "Using S"
#> [1] "function to generate S "
head(quant.p)
#> sum.raw.norm GDI exp.cells
#> 0610007N19Rik 5.158090 2.187929 10.026385
#> 0610007P14Rik 5.354445 1.686117 16.094987
#> 0610009B22Rik 5.366608 1.301230 14.863676
#> 0610009D07Rik 6.805391 1.410419 45.558487
#> 0610009E02Rik 3.677533 1.789017 3.078276
#> 0610009L18Rik 3.260923 1.231797 2.374670In the third column of this data frame is reported the percentage of cells expressing the gene.
NPGs = c("Nes", "Vim", "Sox2", "Sox1", "Notch1",
"Hes1", "Hes5", "Pax6") #,'Hes3'
PNGs = c("Map2", "Tubb3", "Neurod1", "Nefm",
"Nefl", "Dcx", "Tbr1")
hk = c("Calm1", "Cox6b1", "Ppia", "Rpl18",
"Cox7c", "Erh", "H3f3a", "Taf1", "Taf2",
"Gapdh", "Actb", "Golph3", "Mtmr12",
"Zfr", "Sub1", "Tars", "Amacr")
text.size = 12
quant.p$highlight = with(quant.p, ifelse(rownames(quant.p) %in%
NPGs, "NPGs", ifelse(rownames(quant.p) %in%
hk, "Constitutive", ifelse(rownames(quant.p) %in%
PNGs, "PNGs", "normal"))))
textdf <- quant.p[rownames(quant.p) %in%
c(NPGs, hk, PNGs), ]
mycolours <- c(Constitutive = "#00A087FF",
NPGs = "#E64B35FF", PNGs = "#F39B7FFF",
normal = "#8491B4B2")
f1 = ggplot(subset(quant.p, highlight ==
"normal"), aes(x = sum.raw.norm, y = GDI)) +
geom_point(alpha = 0.1, color = "#8491B4B2",
size = 2.5)
GDI_plot = f1 + geom_point(data = subset(quant.p,
highlight != "normal"), aes(x = sum.raw.norm,
y = GDI, colour = highlight), size = 2.5,
alpha = 0.8) + geom_hline(yintercept = quantile(quant.p$GDI)[4],
linetype = "dashed", color = "darkblue") +
geom_hline(yintercept = quantile(quant.p$GDI)[3],
linetype = "dashed", color = "darkblue") +
geom_hline(yintercept = 1.5, linetype = "dotted",
color = "red", size = 0.5) + scale_color_manual("Status",
values = mycolours) + scale_fill_manual("Status",
values = mycolours) + xlab("log normalized counts") +
ylab("GDI") + geom_label_repel(data = textdf,
aes(x = sum.raw.norm, y = GDI, label = rownames(textdf),
fill = highlight), label.size = NA,
alpha = 0.5, direction = "both", na.rm = TRUE,
seed = 1234) + geom_label_repel(data = textdf,
aes(x = sum.raw.norm, y = GDI, label = rownames(textdf)),
label.size = NA, segment.color = "black",
segment.size = 0.5, direction = "both",
alpha = 0.8, na.rm = TRUE, fill = NA,
seed = 1234) + theme(axis.text.x = element_text(size = text.size,
angle = 0, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.text.y = element_text(size = text.size,
angle = 0, hjust = 0, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
axis.title.x = element_text(size = text.size,
angle = 0, hjust = 0.5, vjust = 0,
face = "plain", colour = "#3C5488FF"),
axis.title.y = element_text(size = text.size,
angle = 90, hjust = 0.5, vjust = 0.5,
face = "plain", colour = "#3C5488FF"),
legend.title = element_blank(), legend.text = element_text(color = "#3C5488FF",
face = "italic"), legend.position = "bottom") # titl)
legend <- cowplot::get_legend(GDI_plot)
GDI_plot = GDI_plot + theme(legend.position = "none")
GDI_plot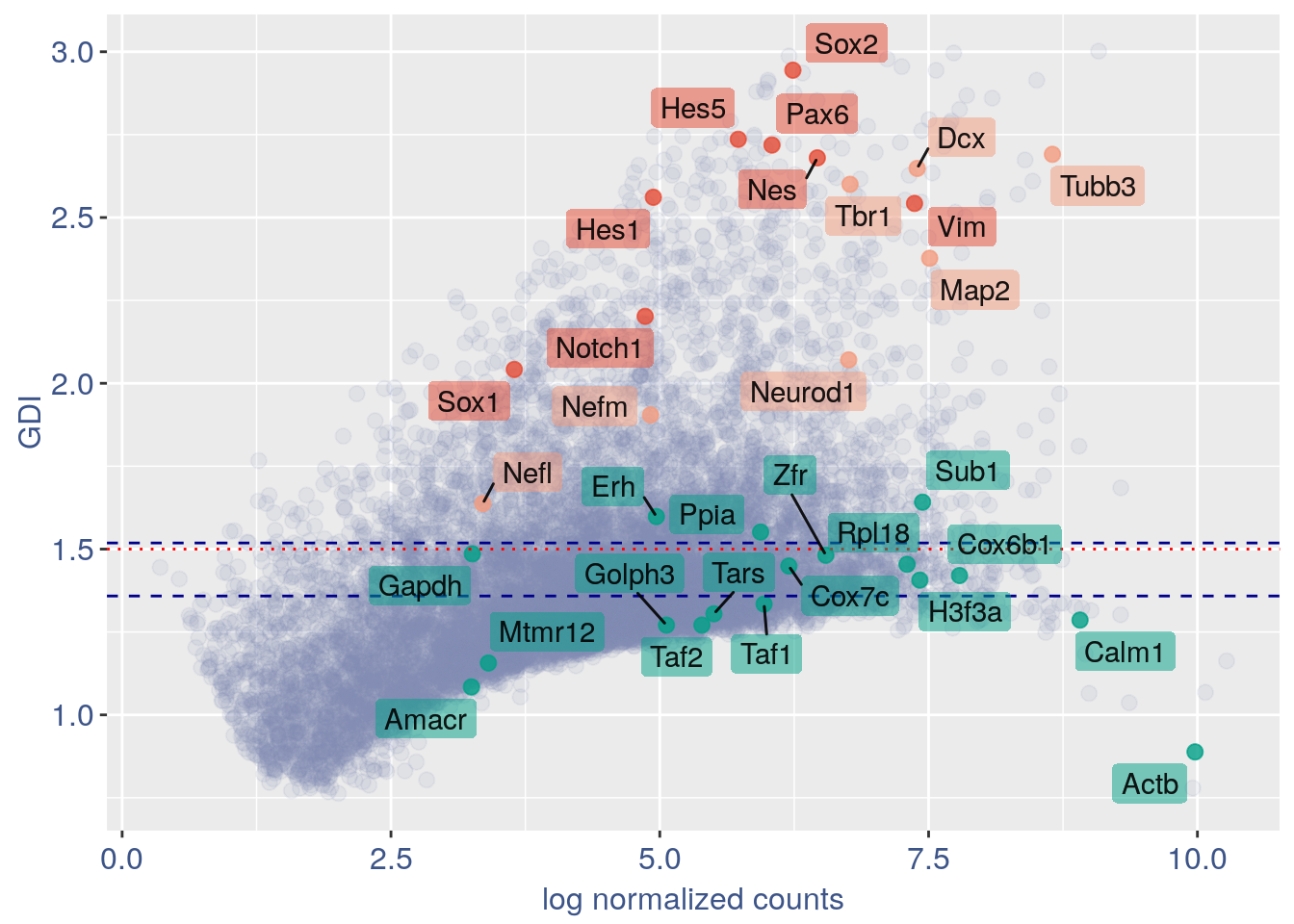
3.3 Heatmaps with cleaning
For the Gene Pair Analysis, we can plot an heatmap with the coex values between two genes sets. To do so we need to define, in a list, the different gene sets (list.genes). Then in the function parameter sets we can decide which sets need to be considered (in the example from 1 to 3). In the condition parameter we should insert an array with each file name prefix to be considered (in the example, the file names is “E13_cortex_clean”).
list.genes = list(Ref.col = PNGs, NPGs = NPGs,
Const. = hk)
plot_heatmap(df_genes = list.genes, sets = c(1:3),
conditions = "E13_cortex_clean", dir = "Data/")
#> [1] "plot heatmap"
#> [1] "Loading condition E13_cortex_clean"
#> [1] "Map2" "Tubb3" "Neurod1" "Nefm" "Nefl" "Dcx" "Tbr1"
#> [1] "Get p-values on a set of genes on columns on a set of genes on rows"
#> [1] "Using function S"
#> [1] "function to generate S "
#> [1] "Ref.col"
#> [1] "NPGs"
#> [1] "Const."
#> [1] "min coex: -0.0121911229941534 max coex 0.00944576864330662"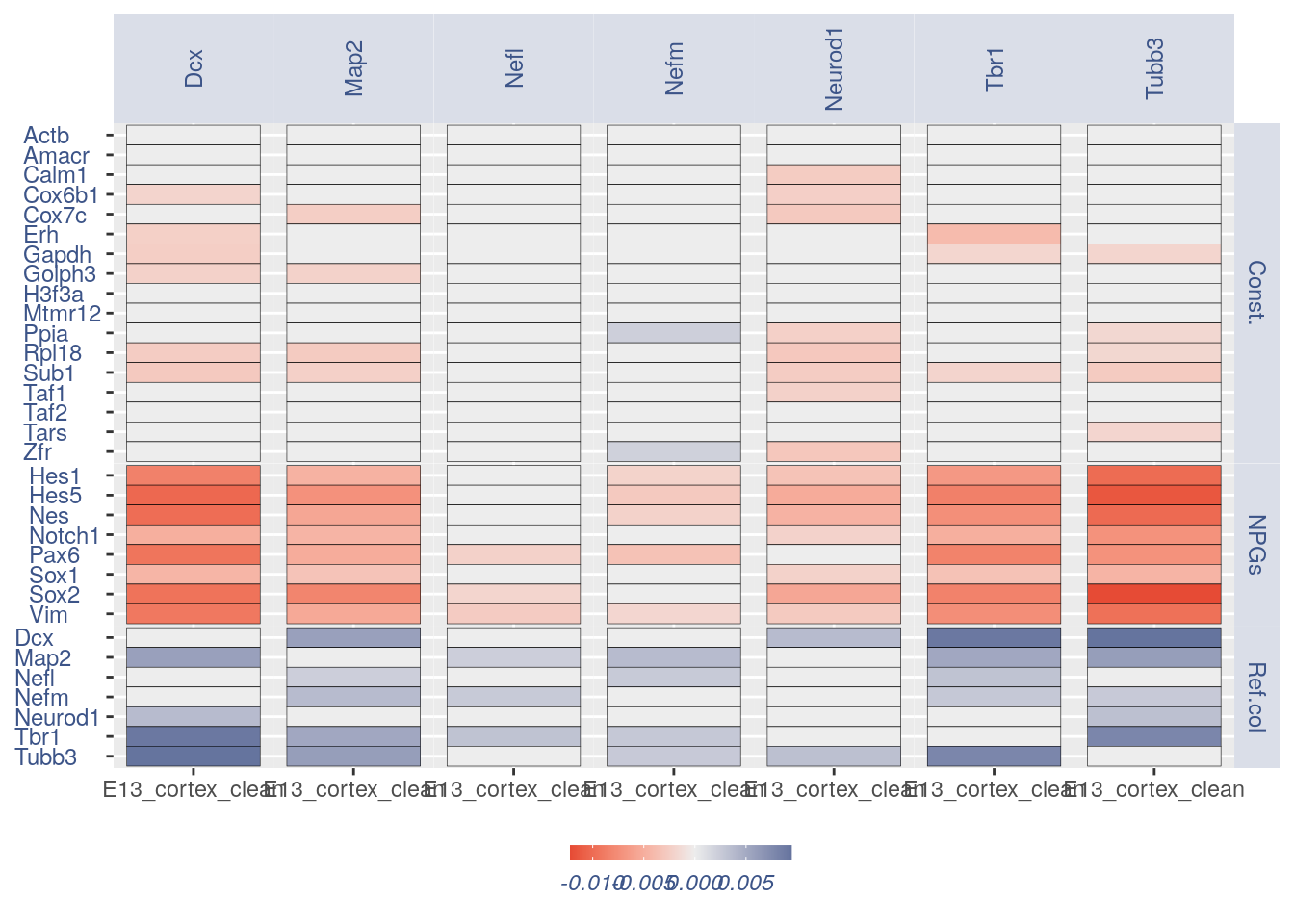
3.4 Heatmaps without cleaning
list.genes = list(Ref.col = PNGs, NPGs = NPGs,
Const. = hk)
plot_heatmap(df_genes = list.genes, sets = c(1:3),
conditions = "E13_cortex", dir = "Data/")
#> [1] "plot heatmap"
#> [1] "Loading condition E13_cortex"
#> [1] "Map2" "Tubb3" "Neurod1" "Nefm" "Nefl" "Dcx" "Tbr1"
#> [1] "Get p-values on a set of genes on columns on a set of genes on rows"
#> [1] "Using function S"
#> [1] "function to generate S "
#> [1] "Ref.col"
#> [1] "NPGs"
#> [1] "Const."
#> [1] "min coex: -0.0120306783883114 max coex 0.00921583325423012"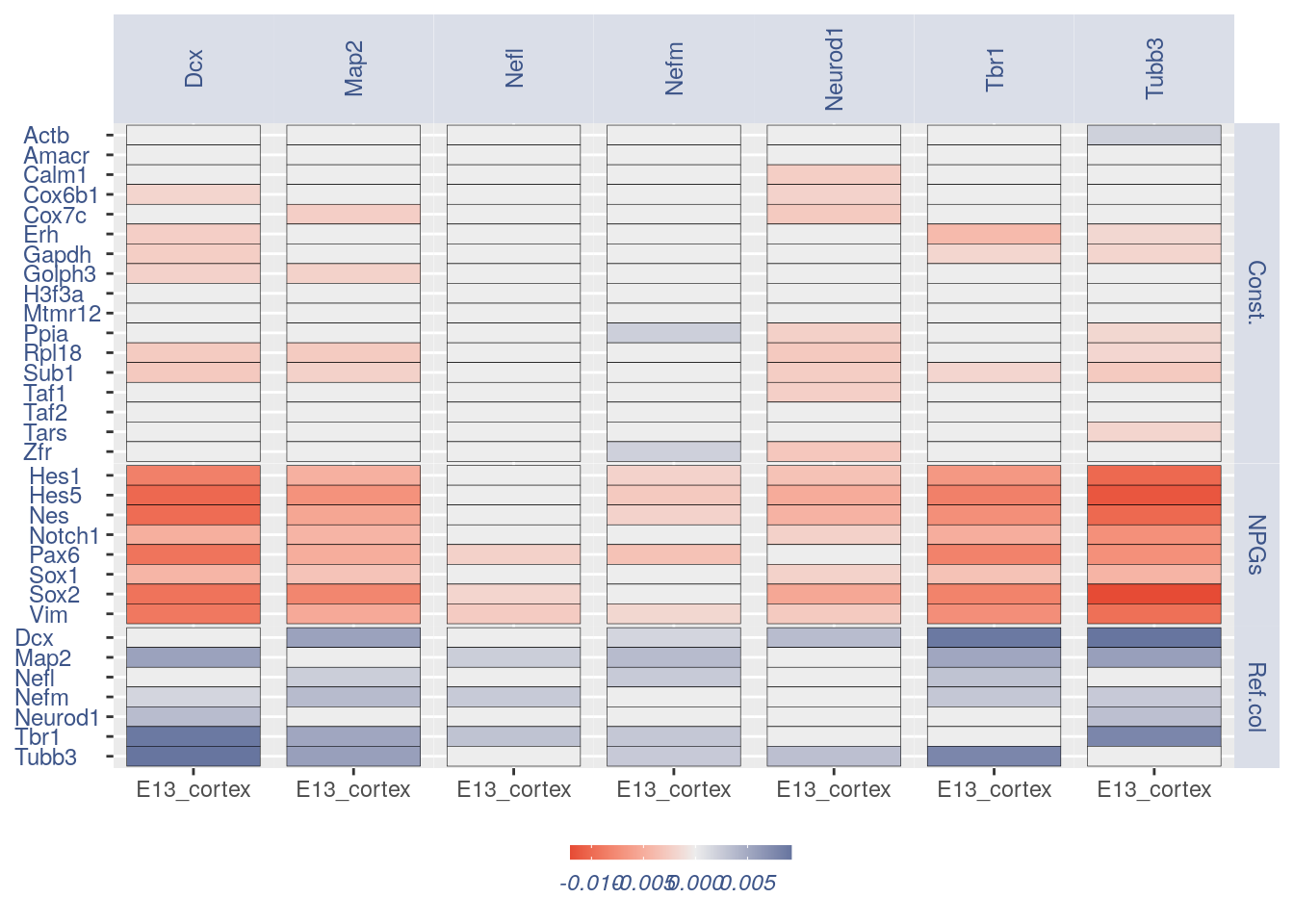
print(sessionInfo())
#> R version 4.0.4 (2021-02-15)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 18.04.5 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/libopenblasp-r0.2.20.so
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggrepel_0.9.1 ggplot2_3.3.3 Matrix_1.3-2 data.table_1.14.0
#> [5] COTAN_0.1.0
#>
#> loaded via a namespace (and not attached):
#> [1] sass_0.3.1 tidyr_1.1.2 jsonlite_1.7.2
#> [4] R.utils_2.10.1 bslib_0.2.4 assertthat_0.2.1
#> [7] highr_0.8 stats4_4.0.4 yaml_2.2.1
#> [10] pillar_1.5.1 lattice_0.20-41 glue_1.4.2
#> [13] reticulate_1.18 digest_0.6.27 RColorBrewer_1.1-2
#> [16] colorspace_2.0-0 cowplot_1.1.1 htmltools_0.5.1.1
#> [19] R.oo_1.24.0 pkgconfig_2.0.3 GetoptLong_1.0.5
#> [22] purrr_0.3.4 scales_1.1.1 tibble_3.1.0
#> [25] generics_0.1.0 farver_2.1.0 IRanges_2.24.1
#> [28] ellipsis_0.3.1 withr_2.4.1 BiocGenerics_0.36.0
#> [31] magrittr_2.0.1 crayon_1.4.0 evaluate_0.14
#> [34] R.methodsS3_1.8.1 fansi_0.4.2 Cairo_1.5-12.2
#> [37] tools_4.0.4 GlobalOptions_0.1.2 formatR_1.8
#> [40] lifecycle_1.0.0 matrixStats_0.58.0 ComplexHeatmap_2.6.2
#> [43] basilisk.utils_1.2.2 stringr_1.4.0 S4Vectors_0.28.1
#> [46] munsell_0.5.0 cluster_2.1.1 compiler_4.0.4
#> [49] jquerylib_0.1.3 rlang_0.4.10 grid_4.0.4
#> [52] rjson_0.2.20 rappdirs_0.3.3 circlize_0.4.12
#> [55] labeling_0.4.2 rmarkdown_2.7 basilisk_1.2.1
#> [58] gtable_0.3.0 DBI_1.1.1 R6_2.5.0
#> [61] knitr_1.31 dplyr_1.0.4 utf8_1.2.1
#> [64] clue_0.3-58 filelock_1.0.2 shape_1.4.5
#> [67] stringi_1.5.3 parallel_4.0.4 Rcpp_1.0.6
#> [70] vctrs_0.3.6 png_0.1-7 tidyselect_1.1.0
#> [73] xfun_0.22