E17.5 cortex
library(COTAN)
library(data.table)
library(Matrix)
library(ggrepel)
#> Loading required package: ggplot2
#library(latex2exp)mycolours <- c("A" = "#8491B4B2","B"="#E64B35FF")
my_theme = theme(axis.text.x = element_text(size = 14, angle = 0, hjust = .5, vjust = .5, face = "plain", colour ="#3C5488FF" ),
axis.text.y = element_text( size = 14, angle = 0, hjust = 0, vjust = .5, face = "plain", colour ="#3C5488FF"),
axis.title.x = element_text( size = 14, angle = 0, hjust = .5, vjust = 0, face = "plain", colour ="#3C5488FF"),
axis.title.y = element_text( size = 14, angle = 90, hjust = .5, vjust = .5, face = "plain", colour ="#3C5488FF"))data = as.data.frame(fread(paste(data_dir,"GSM2861514_E175_Only_Cortical_Cells_DGE.txt.gz", sep = "/")))
data = as.data.frame(data)
rownames(data) = data$V1
data = data[,2:ncol(data)]
data[1:10,1:10]
#> CGTTTAGTTTAC TCTAGAACAACG ACCTTTGTTCGT TTGTCTTCTTCG TAAAATATCGCC
#> 0610005C13Rik 0 0 0 0 0
#> 0610007N19Rik 0 0 0 0 0
#> 0610007P14Rik 2 1 1 1 0
#> 0610009B22Rik 1 1 0 0 0
#> 0610009D07Rik 2 3 0 3 0
#> 0610009E02Rik 0 0 0 0 0
#> 0610009L18Rik 0 0 0 2 0
#> 0610009O20Rik 0 0 0 0 0
#> 0610010F05Rik 1 2 2 0 1
#> 0610010K14Rik 0 0 0 0 0
#> GTACCCTATTTC GCACATTACCCA CCTCGCGCGGCT TTAATTTTGCCT GTCTTGCGTTTT
#> 0610005C13Rik 0 0 0 0 0
#> 0610007N19Rik 0 0 2 0 0
#> 0610007P14Rik 1 0 0 0 0
#> 0610009B22Rik 0 0 1 1 0
#> 0610009D07Rik 2 1 2 1 5
#> 0610009E02Rik 0 0 0 0 0
#> 0610009L18Rik 0 1 1 2 0
#> 0610009O20Rik 0 0 0 1 0
#> 0610010F05Rik 4 0 1 0 2
#> 0610010K14Rik 0 0 0 0 0Define a directory where the ouput will be stored.
1 Analytical pipeline
Initialize the COTAN object with the row count table and the metadata for the experiment.
obj = new("scCOTAN",raw = data)
obj = initRaw(obj,GEO="GSM2861514" ,sc.method="DropSeq",cond = "mouse cortex E17.5")
#> [1] "Initializing S4 object"Now we can start the cleaning. Analysis requires and starts from a matrix of raw UMI counts after removing possible cell doublets or multiplets and low quality or dying cells (with too high mtRNA percentage, easily done with Seurat or other tools).
If we do not want to consider the mitochondrial genes we can remove them before starting the analysis.
genes_to_rem = rownames(obj@raw[grep('^mt', rownames(obj@raw)),]) #genes to remove : mithocondrial
obj@raw = obj@raw[!rownames(obj@raw) %in% genes_to_rem,]
cells_to_rem = colnames(obj@raw[which(colSums(obj@raw) == 0)])
obj@raw = obj@raw[,!colnames(obj@raw) %in% cells_to_rem]We want also to define a prefix to identify the sample.
2 Data cleaning
First, we create the directory to store all information regarding the data cleaning.
if(!file.exists(out_dir)){
dir.create(file.path(out_dir))
}
if(!file.exists(paste(out_dir,"cleaning", sep = ""))){
dir.create(file.path(out_dir, "cleaning"))
}ttm = clean(obj)
#> [1] "Start estimation mu with linear method"
#> [1] 12361 880
#> rowname CCGAGTACGTCC
#> 5083 Hbb-bs 635.916703
#> 5081 Hba-a1 205.857651
#> 5084 Hbb-bt 120.253479
#> 11271 Tuba1a 22.420140
#> 6180 Malat1 18.343751
#> 8461 Ptn 14.267362
#> 4936 Gria2 12.229167
#> 1329 Atrx 10.190973
#> 6820 Mycbp2 8.152778
#> 632 Acin1 6.114584
#> 664 Actb 6.114584
#> 1757 Camk2b 6.114584
#> 1767 Caml 6.114584
#> 5502 Iqsec3 6.114584
#> 8483 Ptprd 6.114584
obj = ttm$object
ttm$pca.cell.2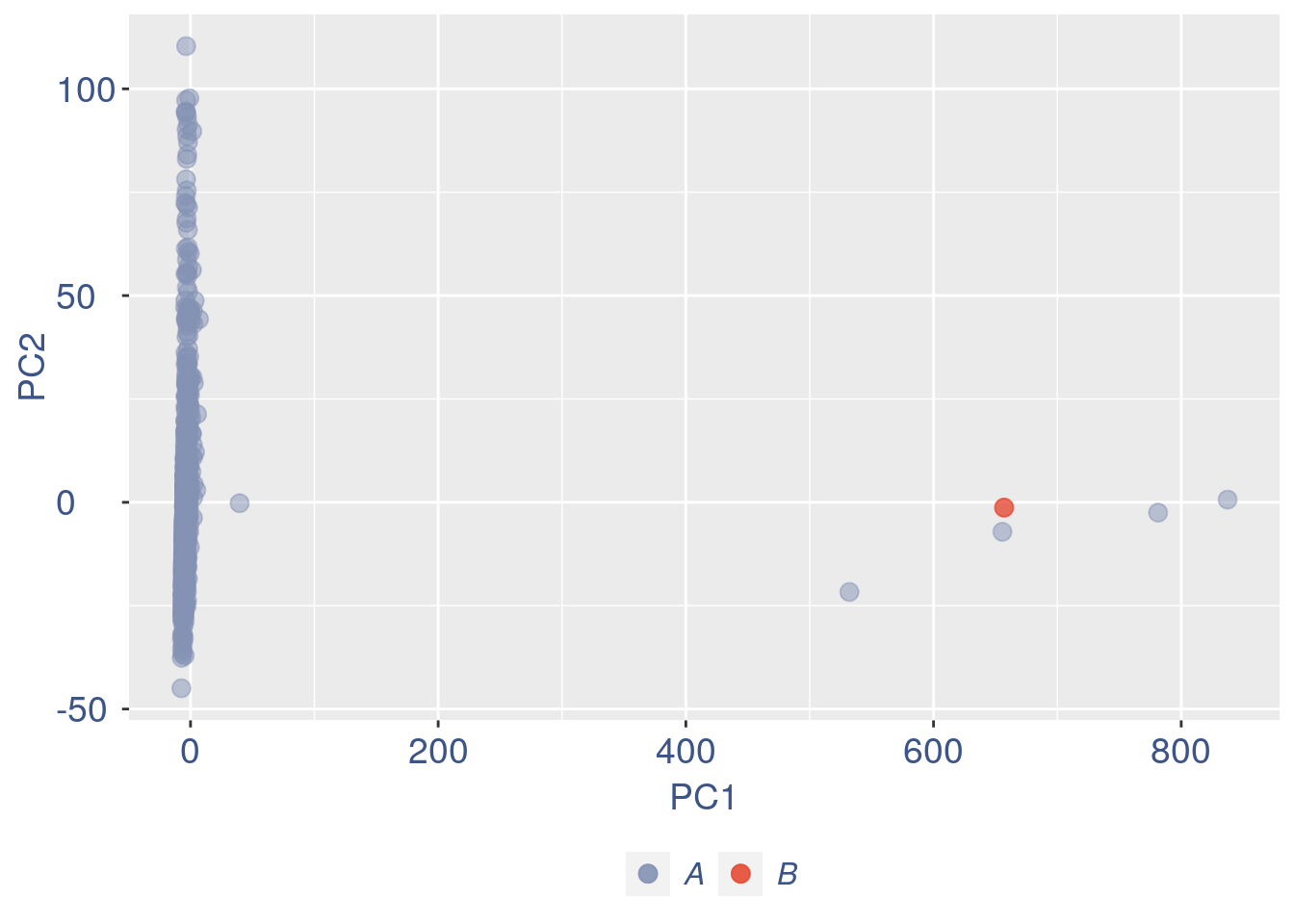
Run this when B cells need to be removed.
pdf(paste(out_dir,"cleaning/",t,"_",n_it,"_plots_before_cells_exlusion.pdf", sep = ""))
ttm$pca.cell.2
ggplot(ttm$D, aes(x=n,y=means)) + geom_point() +
geom_text_repel(data=subset(ttm$D, n > (max(ttm$D$n)- 15) ), aes(n,means,label=rownames(ttm$D[ttm$D$n > (max(ttm$D$n)- 15),])),
nudge_y = 0.05,
nudge_x = 0.05,
direction = "x",
angle = 90,
vjust = 0,
segment.size = 0.2)+
ggtitle("B cell group genes mean expression")+my_theme +
theme(plot.title = element_text(color = "#3C5488FF", size = 20, face = "italic",vjust = - 5,hjust = 0.02 ))
#> Warning: ggrepel: 12 unlabeled data points (too many overlaps). Consider
#> increasing max.overlaps
dev.off()
#> png
#> 2
if (length(ttm$cl1) < length(ttm$cl2)) {
to_rem = ttm$cl1
}else{
to_rem = ttm$cl2
}
n_it = n_it+1
obj@raw = obj@raw[,!colnames(obj@raw) %in% to_rem]
#obj@raw = obj@raw[rownames(obj@raw) %in% rownames(cells),colnames(obj@raw) %in% colnames(cells)]
gc()
#> used (Mb) gc trigger (Mb) max used (Mb)
#> Ncells 3728082 199.2 6227150 332.6 6227150 332.6
#> Vcells 75717375 577.7 118003254 900.3 98268863 749.8
ttm = clean(obj)
#> [1] "Start estimation mu with linear method"
#> [1] 12361 879
#> rowname CGGACATAATGT ACCCGGTCCGAG CACCTGGTCGAC TCGAGTTAAGTC
#> 5083 Hbb-bs 515.0771835 663.4510497 447.1739214 646.150028
#> 5081 Hba-a1 359.0579536 465.9131069 295.5570124 411.900991
#> 5084 Hbb-bt 217.5720219 236.1240715 74.8488538 174.507672
#> 11271 Tuba1a 19.2352475 5.7591237 36.4648262 9.432847
#> 10948 Tmsb4x 4.2744994 1.7277371 21.1112152 1.572141
#> 664 Actb 6.4117492 3.4554742 10.5556076 7.860706
#> 7170 Nnat 12.3960484 6.9109484 0.9596007 3.144282
#> 10944 Tmsb10 4.7019494 0.5759124 12.4748090 4.716424
#> 11279 Tubb2b 3.4195996 2.8795618 9.5960069 3.144282
#> 6395 Meg3 0.4274499 9.7905103 0.0000000 6.288565
#> 11284 Tubb5 5.1293993 1.1518247 6.7172048 3.144282
#> 6180 Malat1 1.2823498 6.3350361 1.9192014 4.716424
#> 1746 Calm1 2.1372497 1.1518247 10.5556076 0.000000
#> 6202 Map1b 2.1372497 1.7277371 6.7172048 3.144282
#> 6065 Lrrc58 2.5646997 1.1518247 4.7980034 4.716424
#ttm = clean.sqrt(obj, cells)
obj = ttm$object
ttm$pca.cell.2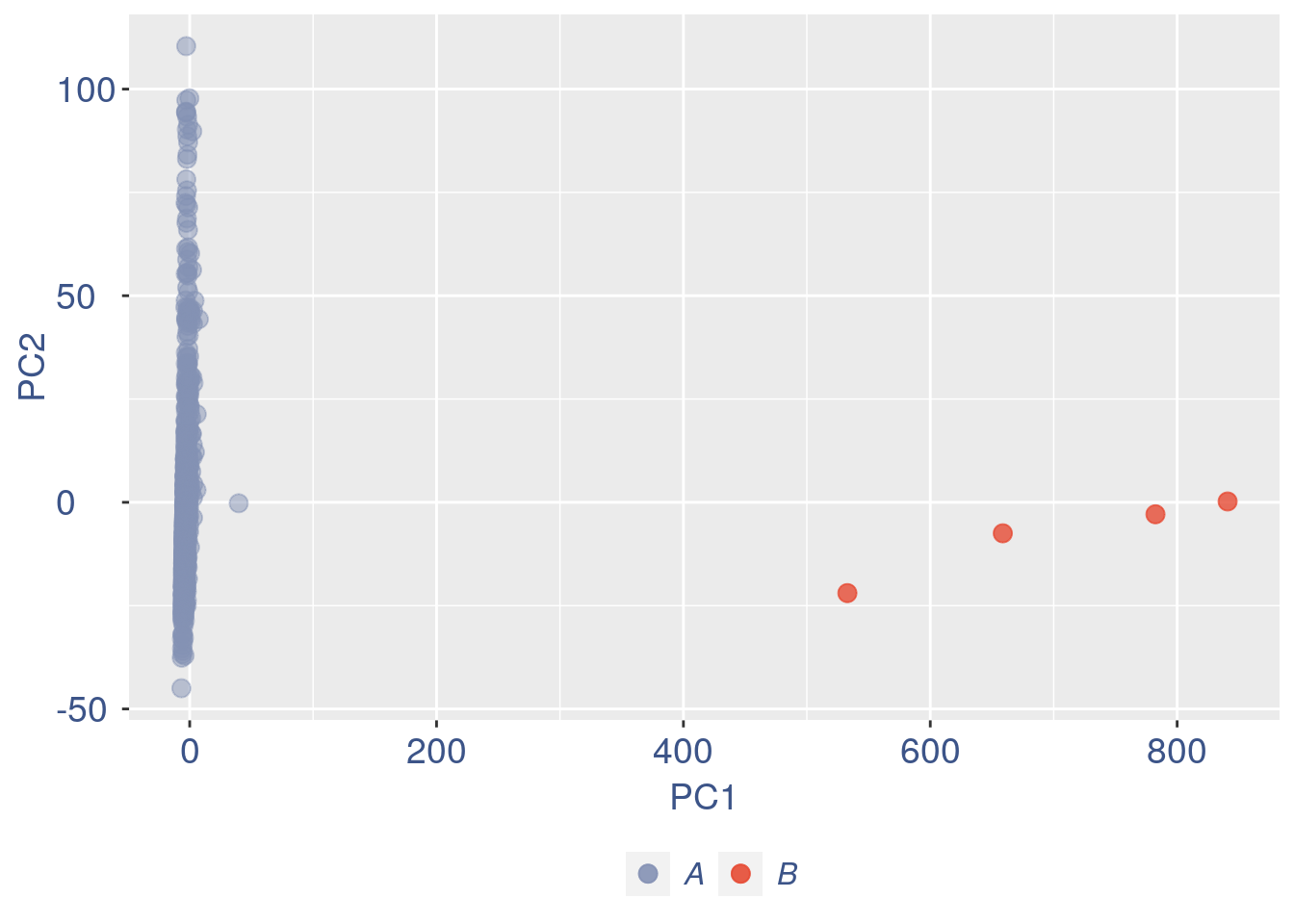
#> Warning: ggrepel: 12 unlabeled data points (too many overlaps). Consider
#> increasing max.overlaps
#> png
#> 2
#> used (Mb) gc trigger (Mb) max used (Mb)
#> Ncells 3729046 199.2 6227150 332.6 6227150 332.6
#> Vcells 77512454 591.4 204200820 1558.0 204200082 1558.0
#> [1] "Start estimation mu with linear method"
#> [1] 12350 875
#> rowname GTGATTGTGTGC CCGGTCAGCAAT TTACTAGCTAAT
#> 6387 Meg3 106.371814 135.402351 134.157606
#> 6173 Malat1 17.406297 37.611764 46.799165
#> 9970 Snhg11 21.274363 56.417646 12.479777
#> 11260 Tuba1a 25.142429 0.000000 18.719666
#> 7162 Nnat 9.670165 30.089411 0.000000
#> 6195 Map1b 15.472264 3.761176 6.239889
#> 9233 Rtn1 11.604198 0.000000 12.479777
#> 11766 Xist 0.000000 0.000000 21.839610
#> 793 Ahi1 1.934033 0.000000 18.719666
#> 9539 Sfrs18 0.000000 11.283529 9.359833
#> 4932 Gria2 5.802099 3.761176 9.359833
#> 11224 Ttc3 5.802099 3.761176 9.359833
#> 7571 Paxbp1 1.934033 3.761176 12.479777
#> 2564 Crmp1 3.868066 7.522353 6.239889
#> 5719 Kif1b 5.802099 11.283529 0.000000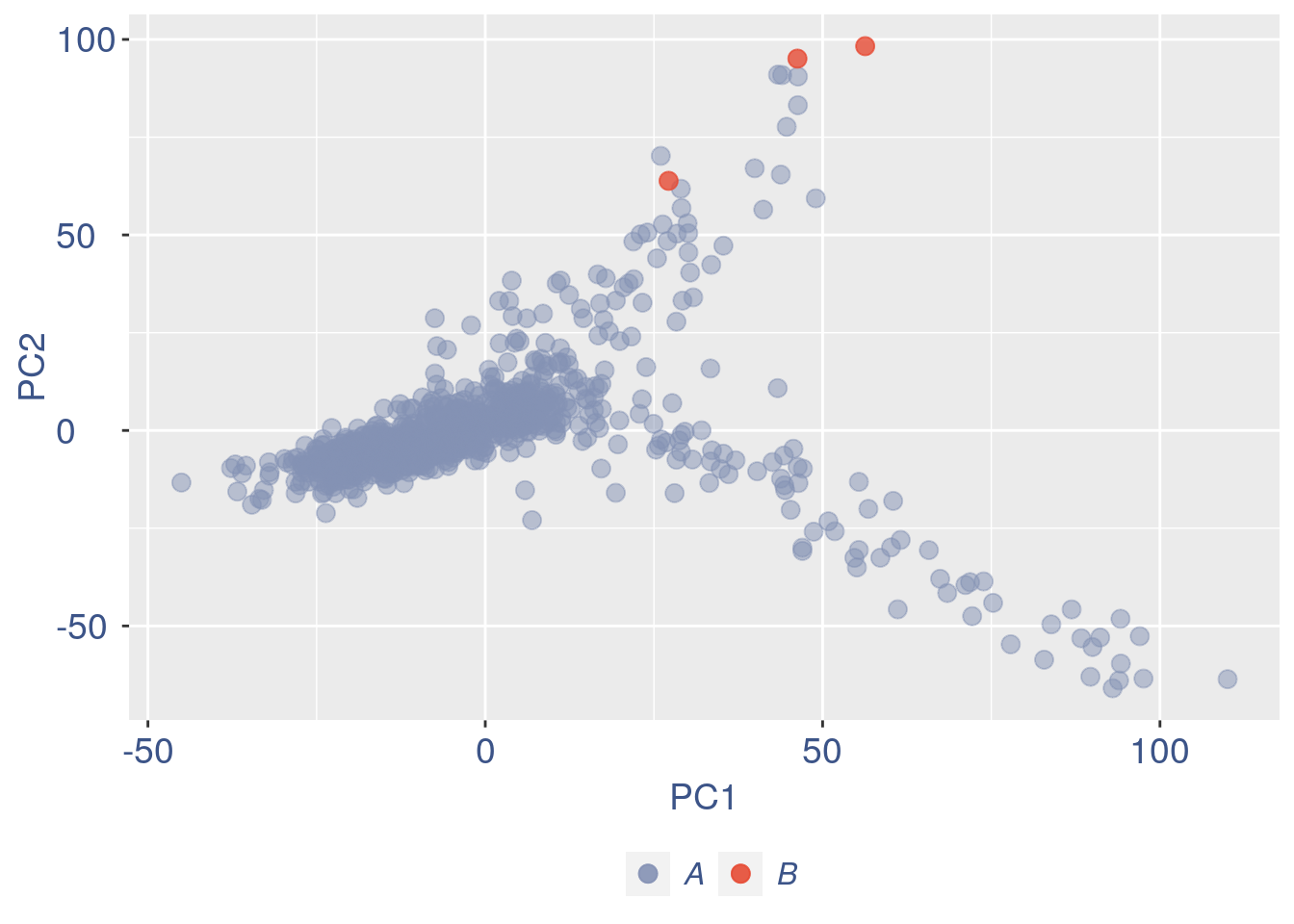
Run this only in the last iteration, instead the previous code, when B cells group has not to be removed.
pdf(paste(out_dir,"cleaning/",t,"_",n_it,"_plots_before_cells_exlusion.pdf", sep = ""))
ttm$pca.cell.2
ggplot(ttm$D, aes(x=n,y=means)) + geom_point() +
geom_text_repel(data=subset(ttm$D, n > (max(ttm$D$n)- 15) ), aes(n,means,label=rownames(ttm$D[ttm$D$n > (max(ttm$D$n)- 15),])),
nudge_y = 0.05,
nudge_x = 0.05,
direction = "x",
angle = 90,
vjust = 0,
segment.size = 0.2)+
ggtitle(label = "B cell group genes mean expression", subtitle = " - B group NOT removed -")+my_theme +
theme(plot.title = element_text(color = "#3C5488FF", size = 20, face = "italic",vjust = - 10,hjust = 0.02 ),
plot.subtitle = element_text(color = "darkred",vjust = - 15,hjust = 0.01 ))
#> Warning: ggrepel: 12 unlabeled data points (too many overlaps). Consider
#> increasing max.overlaps
dev.off()
#> png
#> 2To color the pca based on nu_j (so the cells’ efficiency).
nu_est = round(obj@nu, digits = 7)
plot.nu <-ggplot(ttm$pca_cells,aes(x=PC1,y=PC2, colour = log(nu_est)))
plot.nu = plot.nu + geom_point(size = 1,alpha= 0.8)+
scale_color_gradient2(low = "#E64B35B2",mid = "#4DBBD5B2", high = "#3C5488B2" ,
midpoint = log(mean(nu_est)),name = "ln (nu)")+
ggtitle("Cells PCA coloured by cells efficiency") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF", size = 20),
legend.title=element_text(color = "#3C5488FF", size = 14,face = "italic"),
legend.text = element_text(color = "#3C5488FF", size = 11),
legend.key.width = unit(2, "mm"),
legend.position="right")
pdf(paste(out_dir,"cleaning/",t,"_plots_PCA_efficiency_colored.pdf", sep = ""))
plot.nu
dev.off()
#> png
#> 2
plot.nu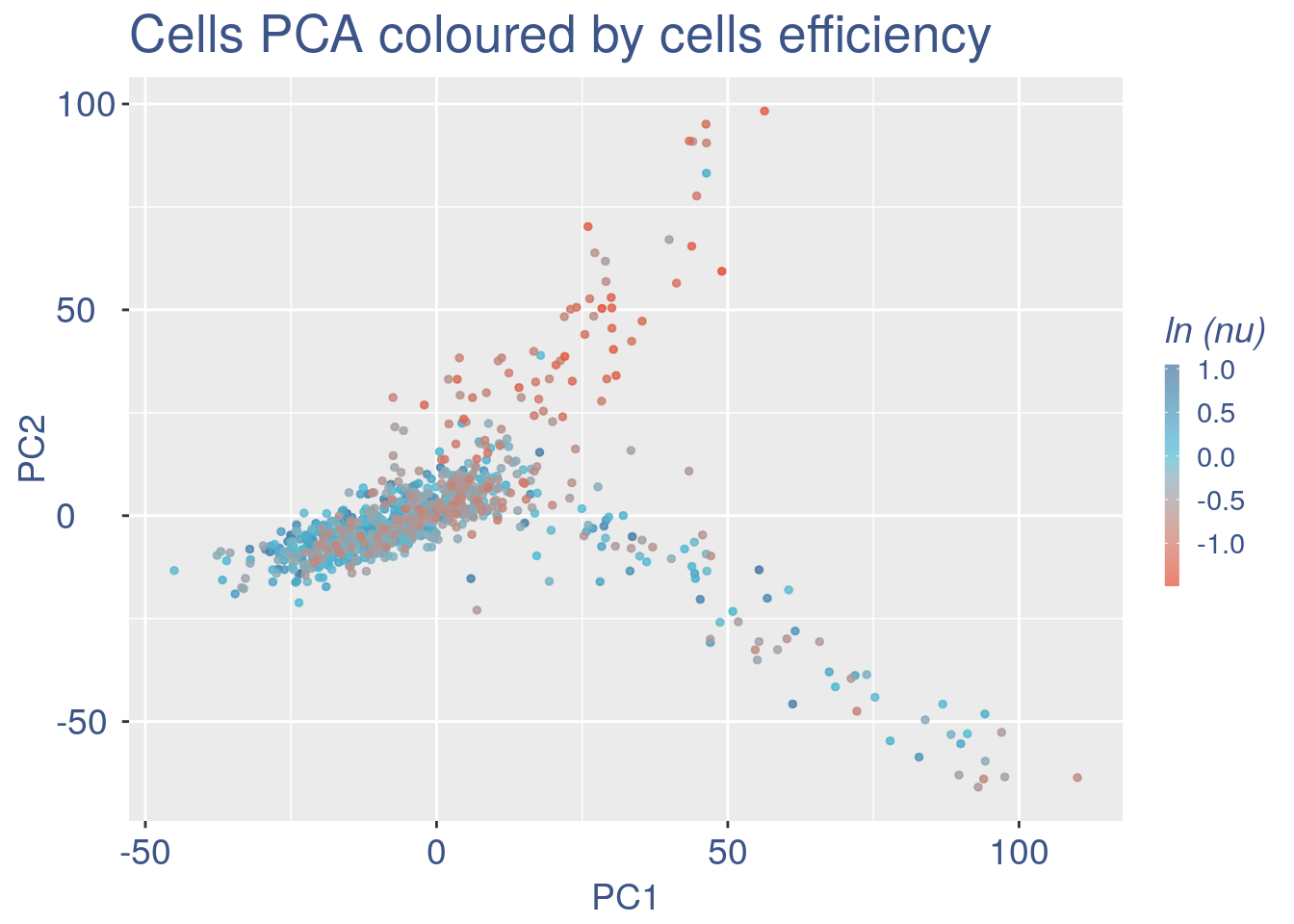
The next part is used to remove the cells with efficiency too low.
nu_df = data.frame("nu"= sort(obj@nu), "n"=c(1:length(obj@nu)))
ggplot(nu_df, aes(x = n, y=nu)) +
geom_point(colour = "#8491B4B2", size=1)+
my_theme #+ ylim(0,1) + xlim(0,70)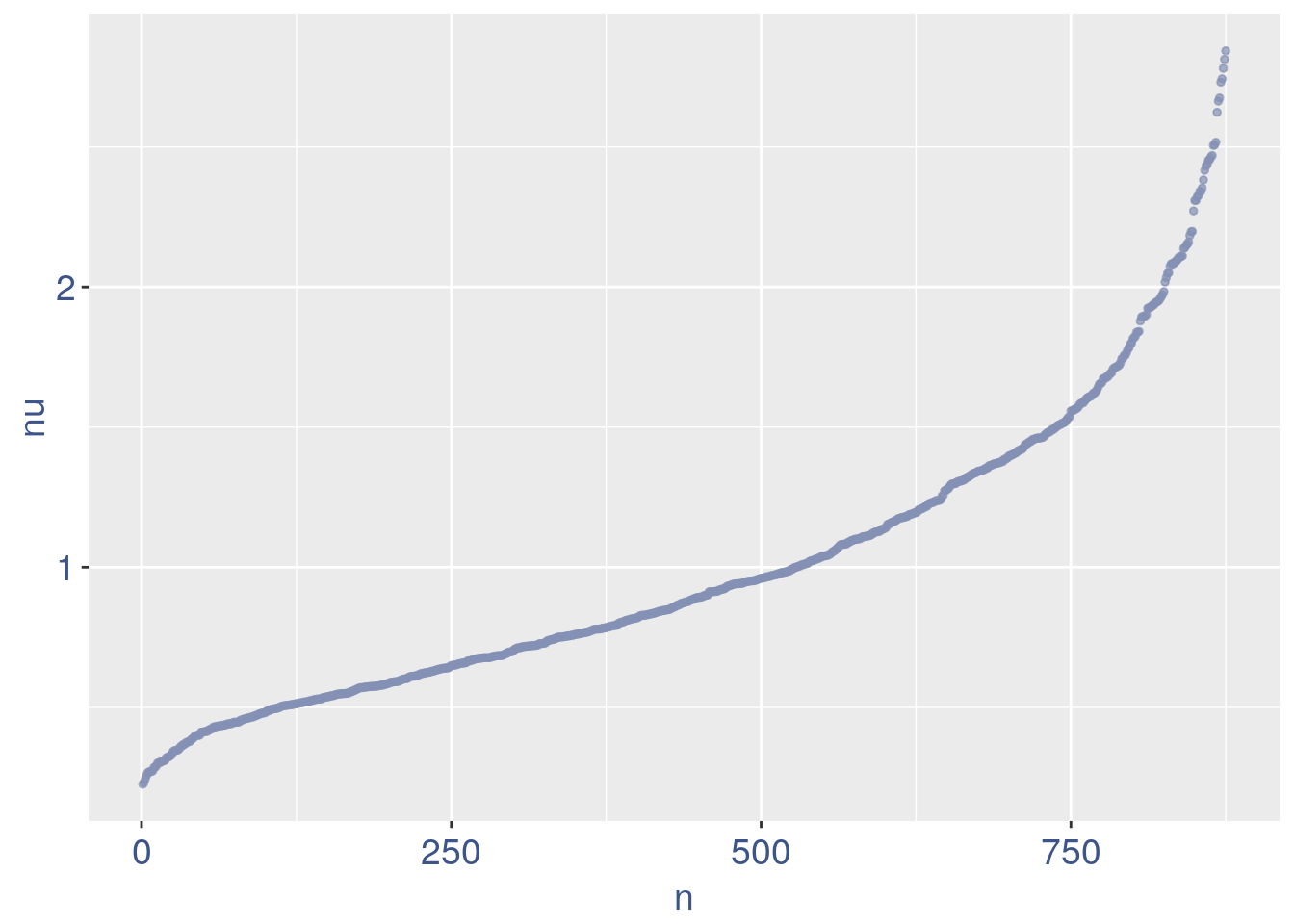
We can zoom on the smallest values and, if we detect a clear elbow, we can decide to remove the cells.
yset = 0.3#threshold to remove low UDE cells
plot.ude <- ggplot(nu_df, aes(x = n, y=nu)) +
geom_point(colour = "#8491B4B2", size=1) +
my_theme + ylim(0.,0.7) + xlim(0,200) +
geom_hline(yintercept=yset, linetype="dashed", color = "darkred") +
annotate(geom="text", x=500, y=0.5,
label=paste("to remove cells with nu < ",yset,sep = " "),
color="darkred", size=4.5)
pdf(paste(out_dir,"cleaning/",t,"_plots_efficiency.pdf", sep = ""))
plot.ude
#> Warning: Removed 675 rows containing missing values (geom_point).
#> Warning: Removed 1 rows containing missing values (geom_text).
dev.off()
#> png
#> 2
plot.ude
#> Warning: Removed 675 rows containing missing values (geom_point).
#> Warning: Removed 1 rows containing missing values (geom_text).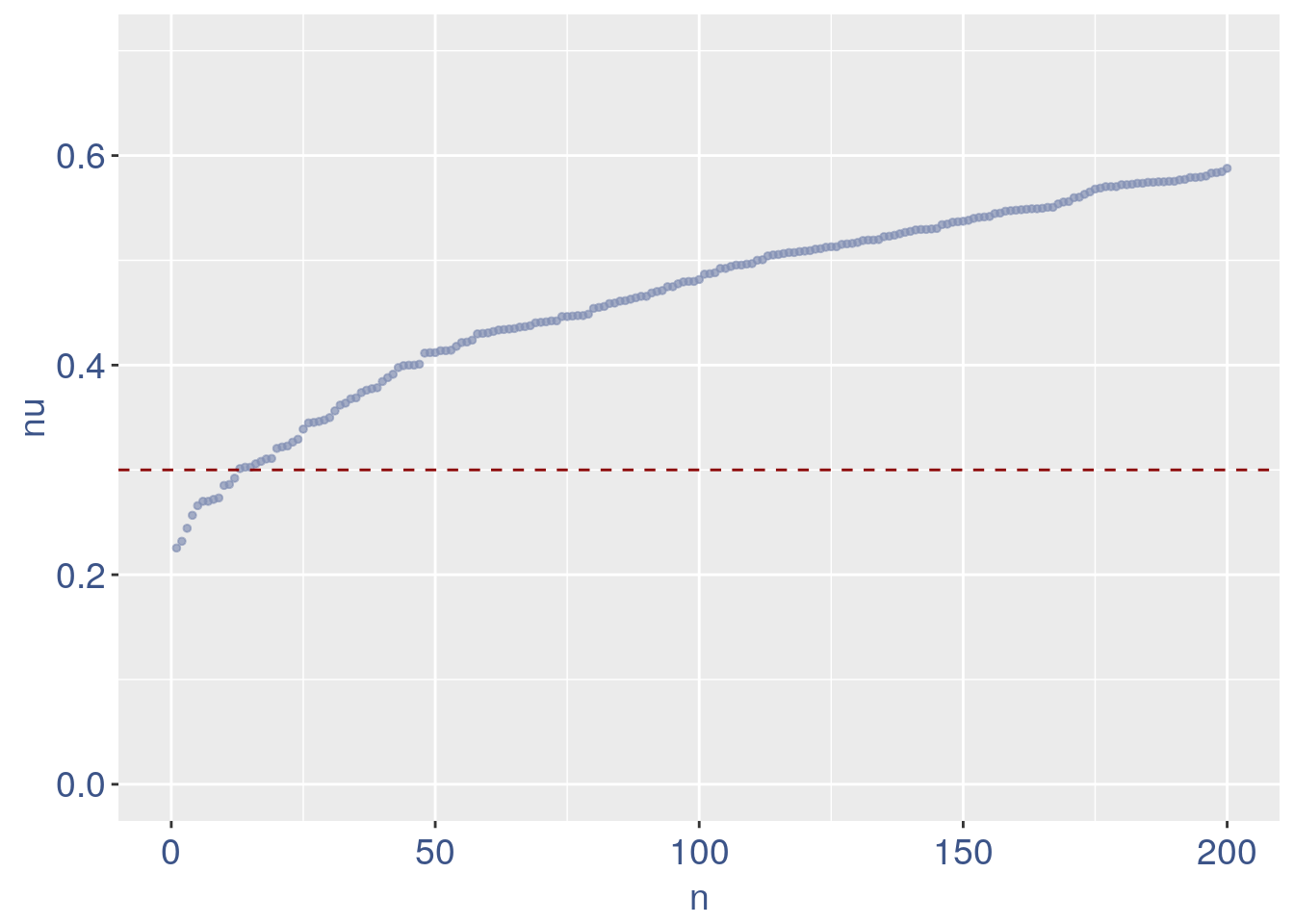
We also save the defined threshold in the metadata and re-run the estimation.
obj@meta[(nrow(obj@meta)+1),1:2] = c("Threshold low UDE cells:",yset)
to_rem = rownames(nu_df[which(nu_df$nu < yset),])
obj@raw = obj@raw[, !colnames(obj@raw) %in% to_rem]Repeat the estimation after the cells are removed.
ttm = clean(obj)
#> [1] "Start estimation mu with linear method"
#> [1] 12335 863
#> rowname GTGATTGTGTGC TTACTAGCTAAT
#> 6379 Meg3 107.457036 135.526303
#> 6166 Malat1 17.583879 47.276617
#> 11246 Tuba1a 25.398936 18.910647
#> 9958 Snhg11 21.491407 12.607098
#> 9222 Rtn1 11.722586 12.607098
#> 11752 Xist 0.000000 22.062421
#> 6188 Map1b 15.630114 6.303549
#> 793 Ahi1 1.953764 18.910647
#> 5241 Hook1 3.907529 12.607098
#> 4926 Gria2 5.861293 9.455323
#> 11210 Ttc3 5.861293 9.455323
#> 7562 Paxbp1 1.953764 12.607098
#> 7403 Olfm1 0.000000 12.607098
#> 9461 Sept3 5.861293 6.303549
#> 8473 Ptprs 11.722586 0.000000
obj = ttm$object
ttm$pca.cell.2
Just to check again, we plot the final efficiency coloured PCA.
nu_est = round(obj@nu, digits = 7)
plot.nu <-ggplot(ttm$pca_cells,aes(x=PC1,y=PC2, colour = log(nu_est)))
plot.nu = plot.nu + geom_point(size = 2,alpha= 0.8)+
scale_color_gradient2(low = "#E64B35B2",mid = "#4DBBD5B2", high = "#3C5488B2" ,
midpoint = log(mean(nu_est)),name = "ln(nu)")+
ggtitle("Cells PCA coloured by cells efficiency: last") +
my_theme + theme(plot.title = element_text(color = "#3C5488FF", size = 20),
legend.title=element_text(color = "#3C5488FF", size = 14,face = "italic"),
legend.text = element_text(color = "#3C5488FF", size = 11),
legend.key.width = unit(2, "mm"),
legend.position="right")
pdf(paste(out_dir,"cleaning/",t,"_plots_PCA_efficiency_colored_FINAL.pdf", sep = ""))
plot.nu
dev.off()
#> png
#> 2
plot.nu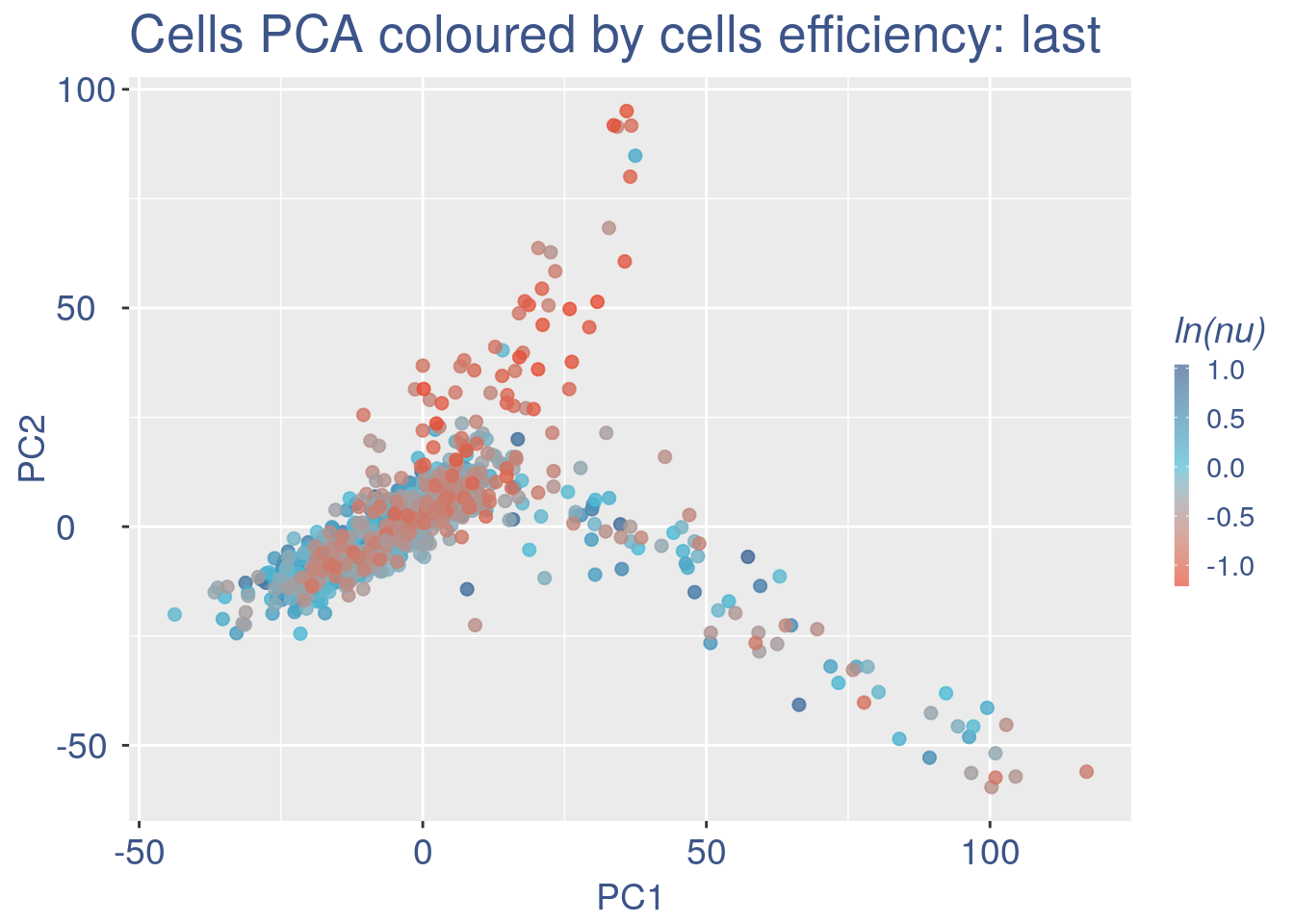
COTAN analysis: in this part all the contingency tables are computed and used to get the statistics (S) To storage efficiency of all the observed tables only the yes_yes is stored. Note that if will be necessary re-computing the yes-yes table, the yes-yes table should be cancelled before running cotan_analysis.
COEX evaluation and storing
3 Analysis of the elaborated data
3.1 GDI
The next function can directly plot the GDI for the dataset with the 1.5 threshold (in red) and the two higher quantiles (in blue).
plot_GDI(obj, cond = "E17.5 cortex")
#> [1] "GDI plot "
#> [1] "function to generate GDI dataframe"
#> [1] "Using S"
#> [1] "function to generate S "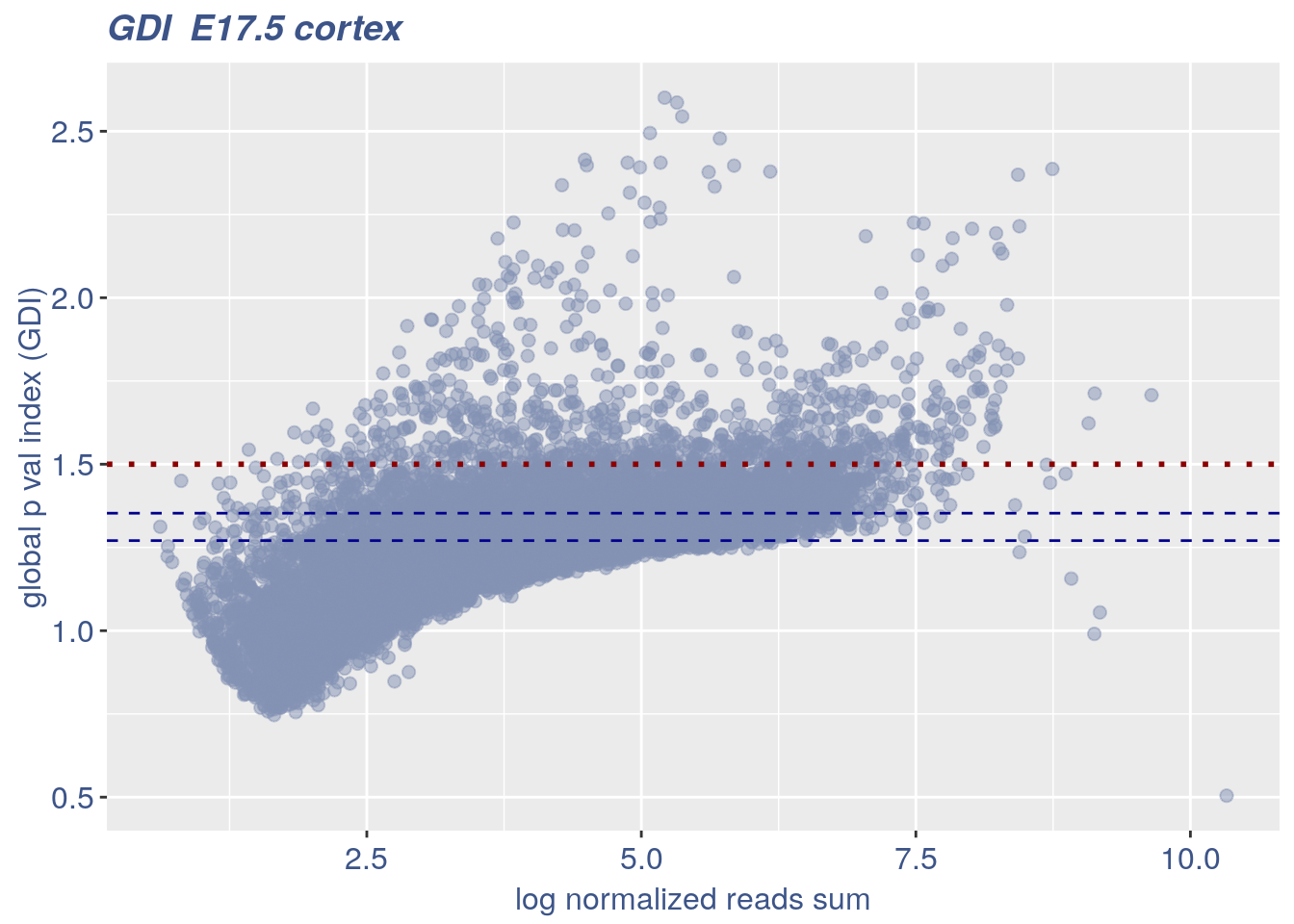
If a more complex plot is needed, or if we want to analyse mor in detail the GDI data, we can get direcly the GDI data frame.
quant.p = get.GDI(obj)
#> [1] "function to generate GDI dataframe"
#> [1] "Using S"
#> [1] "function to generate S "
head(quant.p)
#> sum.raw.norm GDI exp.cells
#> 0610007N19Rik 3.344216 1.609840 2.201622
#> 0610007P14Rik 5.325198 1.380006 19.119351
#> 0610009B22Rik 4.754730 1.293699 11.819235
#> 0610009D07Rik 6.329274 1.354907 43.337196
#> 0610009E02Rik 2.599933 1.007798 1.158749
#> 0610009L18Rik 3.564387 1.342003 3.244496In the third column of this data frame is reported the percentage of cells expressing the gene.
NPGs=c("Nes","Vim","Sox2","Sox1","Notch1", "Hes1","Hes5","Pax6") #,"Hes3"
PNGs=c("Map2","Tubb3","Neurod1","Nefm","Nefl","Dcx","Tbr1")
hk=c("Calm1","Cox6b1","Ppia","Rpl18","Cox7c","Erh","H3f3a","Taf1","Taf2","Gapdh","Actb","Golph3", "Mtmr12", "Zfr", "Sub1", "Tars", "Amacr")
text.size = 12
quant.p$highlight = with(quant.p, ifelse(rownames(quant.p) %in% NPGs, "NPGs",
ifelse(rownames(quant.p) %in% hk,"Constitutive" ,
ifelse(rownames(quant.p) %in% PNGs,"PNGs" , "normal"))))
textdf <- quant.p[rownames(quant.p) %in% c(NPGs,hk,PNGs), ]
mycolours <- c("Constitutive" = "#00A087FF","NPGs"="#E64B35FF","PNGs"="#F39B7FFF","normal" = "#8491B4B2")
f1 = ggplot(subset(quant.p,highlight == "normal" ), aes(x=sum.raw.norm, y=GDI)) + geom_point(alpha = 0.1, color = "#8491B4B2", size=2.5)
GDI_plot = f1 + geom_point(data = subset(quant.p,highlight != "normal" ), aes(x=sum.raw.norm, y=GDI, colour=highlight),size=2.5,alpha = 0.8) +
geom_hline(yintercept=quantile(quant.p$GDI)[4], linetype="dashed", color = "darkblue") +
geom_hline(yintercept=quantile(quant.p$GDI)[3], linetype="dashed", color = "darkblue") +
geom_hline(yintercept=1.5, linetype="dotted", color = "red", size= 0.5) +
scale_color_manual("Status", values = mycolours) +
scale_fill_manual("Status", values = mycolours) +
xlab("log normalized counts")+ylab("GDI")+
geom_label_repel(data =textdf , aes(x=sum.raw.norm, y=GDI, label = rownames(textdf),fill=highlight),
label.size = NA,
alpha = 0.5,
direction ="both",
na.rm=TRUE,
seed = 1234) +
geom_label_repel(data =textdf , aes(x=sum.raw.norm, y=GDI, label = rownames(textdf)),
label.size = NA,
segment.color = 'black',
segment.size = 0.5,
direction = "both",
alpha = 0.8,
na.rm=TRUE,
fill = NA,
seed = 1234) +
theme(axis.text.x = element_text(size = text.size, angle = 0, hjust = .5, vjust = .5, face = "plain", colour ="#3C5488FF" ),
axis.text.y = element_text( size = text.size, angle = 0, hjust = 0, vjust = .5, face = "plain", colour ="#3C5488FF"),
axis.title.x = element_text( size = text.size, angle = 0, hjust = .5, vjust = 0, face = "plain", colour ="#3C5488FF"),
axis.title.y = element_text( size = text.size, angle = 90, hjust = .5, vjust = .5, face = "plain", colour ="#3C5488FF"),
legend.title = element_blank(),
legend.text = element_text(color = "#3C5488FF",face ="italic" ),
legend.position = "bottom") # titl)
legend <- cowplot::get_legend(GDI_plot)
GDI_plot =GDI_plot + theme(
legend.position = "none")
GDI_plot
#> Warning: ggrepel: 1 unlabeled data points (too many overlaps). Consider
#> increasing max.overlaps
#> Warning: ggrepel: 1 unlabeled data points (too many overlaps). Consider
#> increasing max.overlaps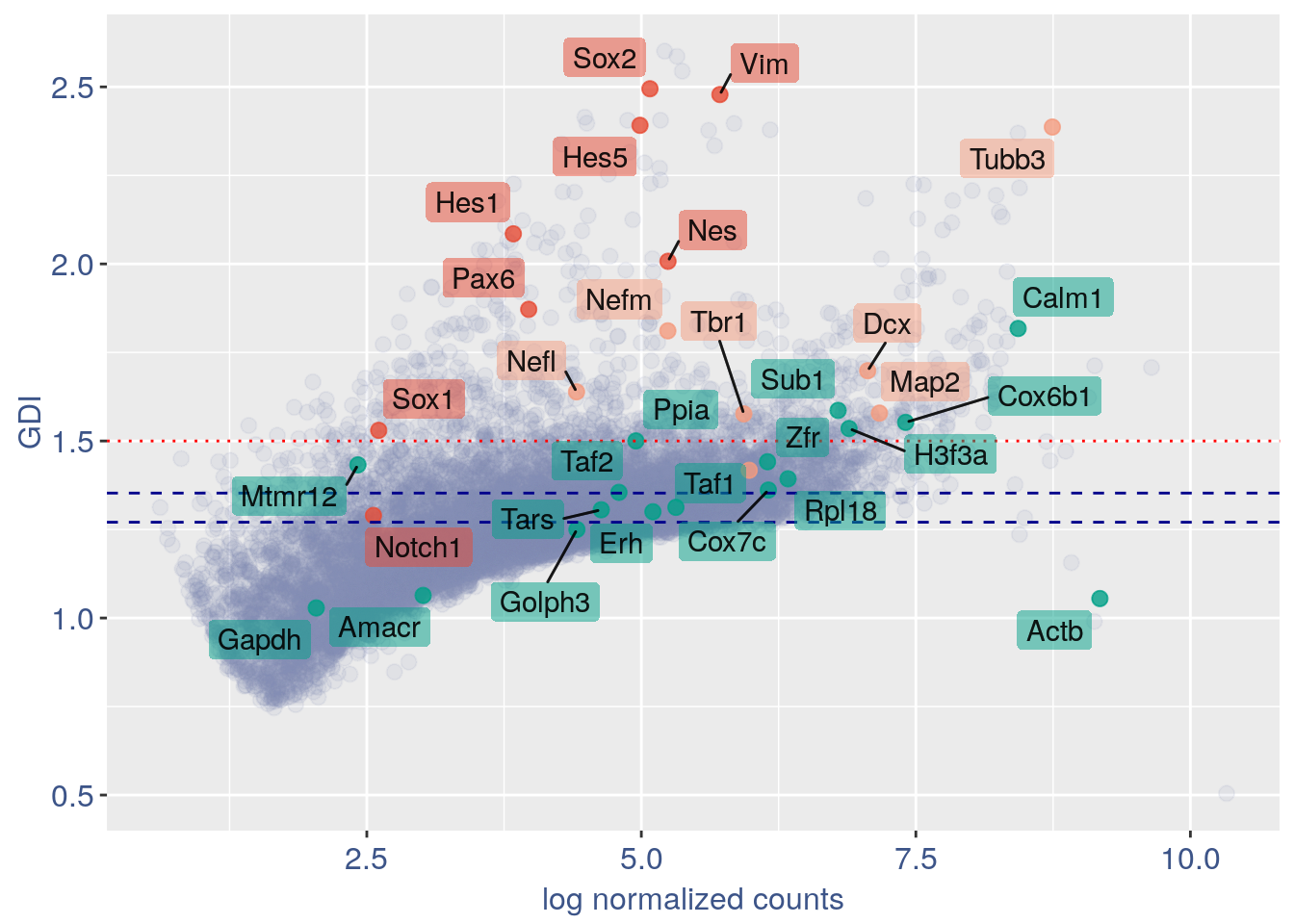
3.2 Heatmaps
For the Gene Pair Analysis, we can plot an heatmap with the coex values between two genes sets. To do so we need to define, in a list, the different gene sets (list.genes). Then in the function parameter sets we can decide which sets need to be considered (in the example from 1 to 3). In the condition parameter we should insert an array with each file name prefix to be considered (in the example, the file names is “E17_cortex_cl2”).
list.genes = list("Ref.col"= PNGs, "NPGs"=NPGs, "Const."=hk )
plot_heatmap(df_genes = list.genes,sets = c(1:3),conditions = "E17_cortex_cl2" ,dir = "Data/")
#> [1] "plot heatmap"
#> [1] "Loading condition E17_cortex_cl2"
#> [1] "Map2" "Tubb3" "Neurod1" "Nefm" "Nefl" "Dcx" "Tbr1"
#> [1] "Get p-values on a set of genes on columns on a set of genes on rows"
#> [1] "Using function S"
#> [1] "function to generate S "
#> [1] "Ref.col"
#> [1] "NPGs"
#> [1] "Const."
#> [1] "min coex: -0.0156874873789823 max coex 0.0114906627057202"
print(sessionInfo())
#> R version 4.0.4 (2021-02-15)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 18.04.5 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/libopenblasp-r0.2.20.so
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggrepel_0.9.1 ggplot2_3.3.3 Matrix_1.3-2 data.table_1.14.0
#> [5] COTAN_0.1.0
#>
#> loaded via a namespace (and not attached):
#> [1] Rcpp_1.0.6 lattice_0.20-41 circlize_0.4.12
#> [4] tidyr_1.1.2 png_0.1-7 assertthat_0.2.1
#> [7] digest_0.6.27 utf8_1.2.1 R6_2.5.0
#> [10] stats4_4.0.4 evaluate_0.14 highr_0.8
#> [13] pillar_1.5.1 basilisk_1.2.1 GlobalOptions_0.1.2
#> [16] rlang_0.4.10 jquerylib_0.1.3 R.oo_1.24.0
#> [19] R.utils_2.10.1 S4Vectors_0.28.1 GetoptLong_1.0.5
#> [22] reticulate_1.18 rmarkdown_2.7 labeling_0.4.2
#> [25] stringr_1.4.0 munsell_0.5.0 compiler_4.0.4
#> [28] xfun_0.22 pkgconfig_2.0.3 BiocGenerics_0.36.0
#> [31] shape_1.4.5 htmltools_0.5.1.1 tidyselect_1.1.0
#> [34] tibble_3.1.0 IRanges_2.24.1 matrixStats_0.58.0
#> [37] fansi_0.4.2 withr_2.4.1 crayon_1.4.0
#> [40] dplyr_1.0.4 R.methodsS3_1.8.1 rappdirs_0.3.3
#> [43] basilisk.utils_1.2.2 grid_4.0.4 jsonlite_1.7.2
#> [46] gtable_0.3.0 lifecycle_1.0.0 DBI_1.1.1
#> [49] magrittr_2.0.1 scales_1.1.1 stringi_1.5.3
#> [52] farver_2.1.0 bslib_0.2.4 ellipsis_0.3.1
#> [55] filelock_1.0.2 generics_0.1.0 vctrs_0.3.6
#> [58] cowplot_1.1.1 rjson_0.2.20 RColorBrewer_1.1-2
#> [61] tools_4.0.4 Cairo_1.5-12.2 glue_1.4.2
#> [64] purrr_0.3.4 parallel_4.0.4 yaml_2.2.1
#> [67] clue_0.3-58 colorspace_2.0-0 cluster_2.1.1
#> [70] ComplexHeatmap_2.6.2 knitr_1.31 sass_0.3.1