Loo et all markers
library(COTAN)
library(ggplot2)
library(data.table)
library(Matrix)
library(factoextra)
#> Welcome! Want to learn more? See two factoextra-related books at https://goo.gl/ve3WBa
library(ggrepel)
library(dendextend)
#>
#> ---------------------
#> Welcome to dendextend version 1.14.0
#> Type citation('dendextend') for how to cite the package.
#>
#> Type browseVignettes(package = 'dendextend') for the package vignette.
#> The github page is: https://github.com/talgalili/dendextend/
#>
#> Suggestions and bug-reports can be submitted at: https://github.com/talgalili/dendextend/issues
#> Or contact: <tal.galili@gmail.com>
#>
#> To suppress this message use: suppressPackageStartupMessages(library(dendextend))
#> ---------------------
#>
#> Attaching package: 'dendextend'
#> The following object is masked from 'package:data.table':
#>
#> set
#> The following object is masked from 'package:stats':
#>
#> cutree1 Gene clustering using COTAN
input_dir = "Data/"
layers = list("L1"=c("Reln","Lhx5"), "L2/3"=c("Satb2","Cux1"), "L4"=c("Rorb","Sox5") , "L5/6"=c("Bcl11b","Fezf2") , "Prog"=c("Vim","Hes1"))
objE17 = readRDS(file = "Data/E17_cortex_cl2.cotan.RDS")g.space = get.gene.coexpression.space(objE17,
n.genes.for.marker = 25, primary.markers = unlist(layers))
#> [1] "calculating gene coexpression space: output tanh of reduced coex matrix"
#> L11 L12 L2/31 L2/32 L41 L42 L5/61 L5/62
#> "Reln" "Lhx5" "Satb2" "Cux1" "Rorb" "Sox5" "Bcl11b" "Fezf2"
#> Prog1 Prog2
#> "Vim" "Hes1"
#> [1] "Get p-values on a set of genes on columns genome wide on rows"
#> [1] "Using function S"
#> [1] "function to generate S "
#> [1] "Secondary markers:181"
#> [1] "function to generate S "
#> [1] "Columns (V set) number: 181 Rows (U set) number: 1236"g.space = as.data.frame(as.matrix(g.space))
coex.pca.genes <- prcomp(t(g.space),
center = TRUE,
scale. = F)
fviz_eig(coex.pca.genes, addlabels=TRUE,ncp = 10)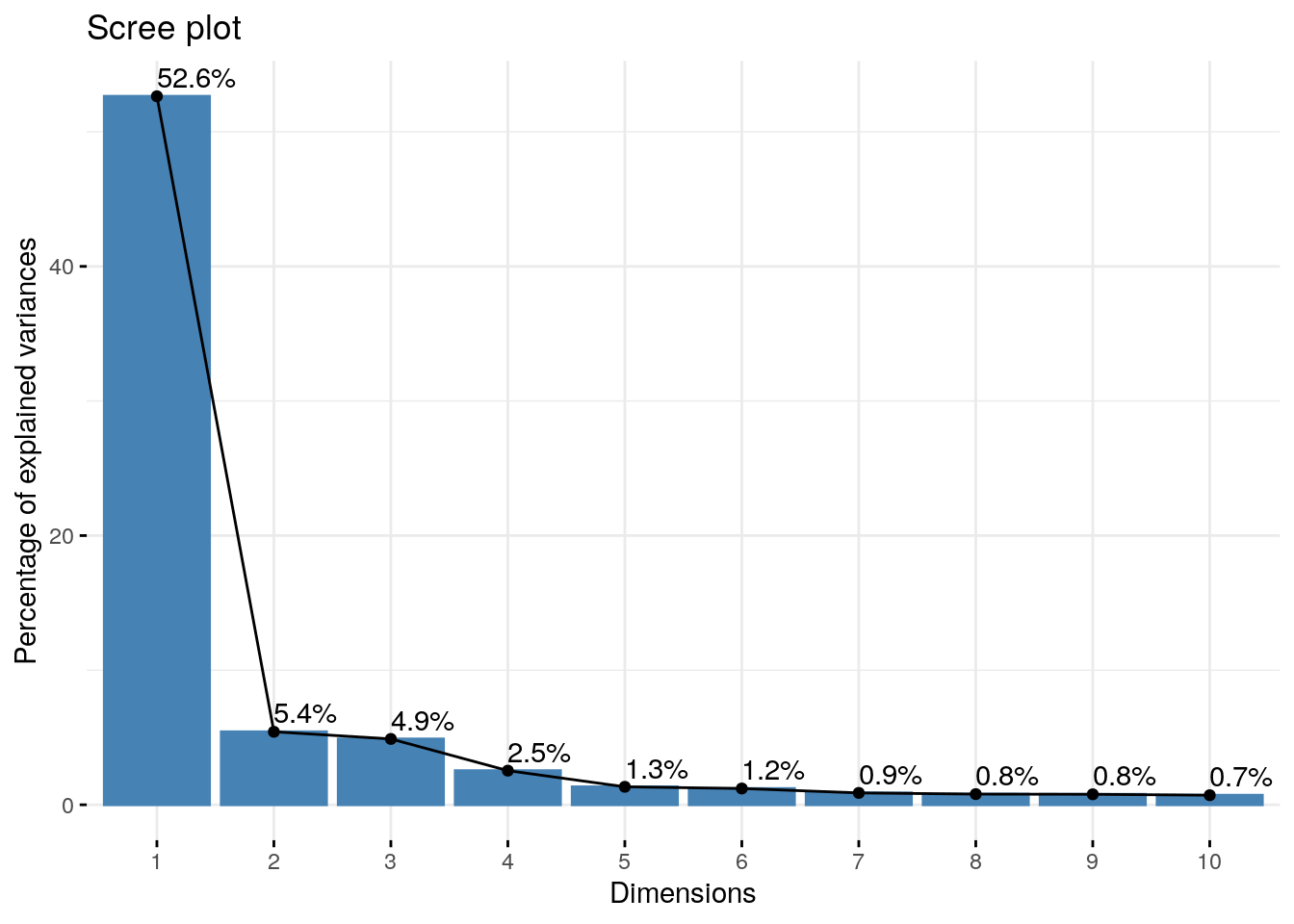
1.1 Hierarchical clustering
controls =list("genes related to L5/6"=c("Foxp2","Tbr1"), "genes related to L2/3"=c("Mef2c"), "genes related to Prog"=c("Nes","Sox2") , "genes related to L1"=c() , "genes related to L4"=c())
# clustering usign Ward method
hc.norm = hclust(dist(g.space), method = "ward.D2")
# and cut the tree into 5 clusters (for example)
cut = cutree(hc.norm, k = 7, order_clusters_as_data = F)
# It crates the tree
dend <- as.dendrogram(hc.norm)
# I can use a dataframe from the pca to store some data regarding the clustering
pca_1 = as.data.frame(coex.pca.genes$rotation[,1:5])
pca_1 = pca_1[order.dendrogram(dend),]
mycolours <- c("genes related to L5/6" = "#3C5488FF","genes related to L2/3"="#F39B7FFF","genes related to Prog"="#4DBBD5FF","genes related to L1"="#E64B35FF","genes related to L4" = "#91D1C2FF", "not marked"="#B09C85FF")
# save the cluster number into the dataframe
pca_1$hclust = cut
dend =branches_color(dend,k=7,col=c("#4DBBD5FF","#91D1C2FF","#F39B7FFF","#E64B35FF","#3C5488FF","#91D1C2FF","#B09C85FF" ),groupLabels = T)
#dend =color_labels(dend,k=5,labels = rownames(pca_1),col=pca_1$colors)
dend %>%
set("labels", ifelse(labels(dend) %in% rownames(pca_1)[rownames(pca_1) %in% c(unlist(layers),unlist(controls))], labels(dend), "")) %>%
set("branches_k_color", value = c("gray80","#4DBBD5FF","#91D1C2FF" ,"gray80","#F39B7FFF","#E64B35FF","#3C5488FF"), k = 7) %>%
plot(horiz=F, axes=T,ylim = c(0,100))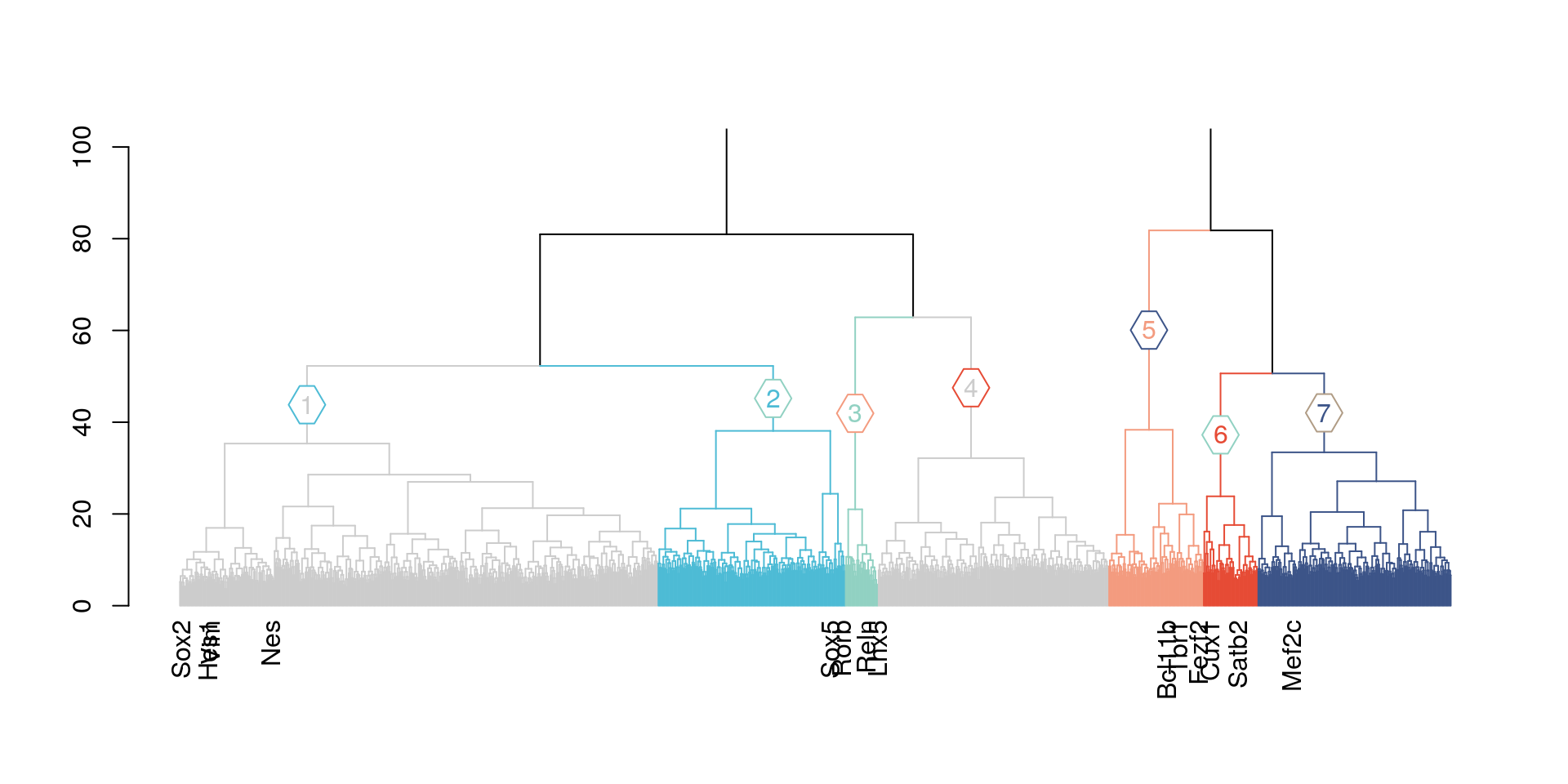
1.2 Import of markers from Loo et al. paper
Markers_Loo = read.csv("Data/Markers_Loo.csv")
genes_detected = pca_1[rownames(pca_1) %in% unlist(Markers_Loo)[!unlist(Markers_Loo) == ""],]Gene present in the 10% of most differentially expressed genes by COTAN
of total genes detected as markers by Loo et al
Number of genes detected:
Removed becouse not detected
pure.markers = unlist(Markers_Loo)
pure.markers = pure.markers[!unlist(Markers_Loo) == ""]
pure.markers[!pure.markers %in% rownames(objE17@coex)]
#> L.I2 PROG4 PROG10 PROG19 PROG22
#> "Gdf5" "Cdc25c" "Gas2l3" "Rspo1" "Wnt8b"Markers_Loo = as.list(Markers_Loo)
for (i in names(Markers_Loo)) {
Markers_Loo[[i]] = Markers_Loo[[i]][! Markers_Loo[[i]] == ""]
}
Markers_Loo
#> $L.I
#> [1] "Ebf3" "Gdf5" "Lhx1" "Lhx5" "Ndnf" "Reln" "Samd3" "Trp73"
#>
#> $L.II.IV
#> [1] "Satb2" "3110047P20Rik" "9130024F11Rik" "Dok5"
#> [5] "Inhba" "Pou3f1"
#>
#> $L.V.VI
#> [1] "Bcl11b" "Crym" "Fezf2" "Hs3st4" "Mc4r" "Nfe2l3" "Nxph3" "Plxna4"
#> [9] "Rwdd3" "Sla" "Sybu" "Tbr1"
#>
#> $PROG
#> [1] "Aldoc" "Arhgef39" "Aspm" "Cdc25c" "Cdkn3" "Cyr61"
#> [7] "Dkk3" "Ednrb" "Gas1" "Gas2l3" "Hes1" "Hes5"
#> [13] "Htra1" "Nde1" "Nek2" "Pax6" "Pkmyt1" "Plk1"
#> [19] "Rspo1" "Tcf19" "Tk1" "Wnt8b"Primary markers also used by Loo et al.
unlist(layers)[unlist(layers) %in% unique(unlist(Markers_Loo))]
#> L11 L12 L2/31 L5/61 L5/62 Prog2
#> "Reln" "Lhx5" "Satb2" "Bcl11b" "Fezf2" "Hes1"Table
tableMarkersCOTAN = as.data.frame(matrix(nrow = 6,ncol = 4))
colnames(tableMarkersCOTAN)=c("Loo.L.I","Loo.L.II.IV","Loo.L.V.VI","Loo.PROG")
rownames(tableMarkersCOTAN)=c("COTAN.L.I","COTAN.L.II.III","COTAN.L.IV","COTAN.L.V.VI","COTAN.PROG","COTAN.Not Grouped")
tableMarkersCOTAN_no_sec = tableMarkersCOTAN
layers.cluster = c("Reln","Satb2","Rorb","Bcl11b","Vim")
names(layers.cluster) = unique(cut[unlist(layers)])
layers.cluster
#> 3 6 2 5 1
#> "Reln" "Satb2" "Rorb" "Bcl11b" "Vim"1.3 Comparition between Loo et al. markers and COTAN markers
groups = list("L.I"=names(which(layers.cluster == "Reln"))
,"L.II.III"=names(which(layers.cluster == "Satb2")),
"L.IV"=names(which(layers.cluster == "Rorb")),
"L.V.VI"=names(which(layers.cluster == "Bcl11b")),
"PROG"=names(which(layers.cluster == "Vim")))
for(layer1 in c("L.I","L.II.III", "L.IV","L.V.VI","PROG")){
for(layer2 in c("L.I","L.II.IV","L.V.VI","PROG")){
tableMarkersCOTAN[paste0("COTAN.",layer1),paste0("Loo.",layer2)] =
sum(rownames(pca_1[pca_1$hclust %in% groups[[layer1]],]) %in% Markers_Loo[[layer2]])
tableMarkersCOTAN[paste0("COTAN.","Not Grouped"),paste0("Loo.",layer2)] =
sum(rownames(pca_1[!pca_1$hclust %in% unlist(groups),]) %in% Markers_Loo[[layer2]])
}
}
tableMarkersCOTAN
#> Loo.L.I Loo.L.II.IV Loo.L.V.VI Loo.PROG
#> COTAN.L.I 6 0 0 0
#> COTAN.L.II.III 0 4 1 0
#> COTAN.L.IV 0 0 0 1
#> COTAN.L.V.VI 0 0 9 0
#> COTAN.PROG 0 0 0 9
#> COTAN.Not Grouped 0 0 1 2Without secondary markers
pca_2 = pca_1[! rownames(pca_1) %in% colnames(g.space),]
for(layer1 in c("L.I","L.II.III", "L.IV","L.V.VI","PROG")){
for(layer2 in c("L.I","L.II.IV","L.V.VI","PROG")){
tableMarkersCOTAN_no_sec[paste0("COTAN.",layer1),paste0("Loo.",layer2)] =
sum(rownames(pca_2[pca_2$hclust %in% groups[[layer1]],]) %in% Markers_Loo[[layer2]])
tableMarkersCOTAN_no_sec[paste0("COTAN.","Not Grouped"),paste0("Loo.",layer2)] =
sum(rownames(pca_2[!pca_2$hclust %in% unlist(groups),]) %in% Markers_Loo[[layer2]])
}
}
tableMarkersCOTAN_no_sec
#> Loo.L.I Loo.L.II.IV Loo.L.V.VI Loo.PROG
#> COTAN.L.I 0 0 0 0
#> COTAN.L.II.III 0 2 1 0
#> COTAN.L.IV 0 0 0 1
#> COTAN.L.V.VI 0 0 5 0
#> COTAN.PROG 0 0 0 5
#> COTAN.Not Grouped 0 0 1 2specific.genes.table = data.frame("genes"=c(), "COTAN"=c(),"Loo."=c())
tt1 = c()
tt2 = c()
for(layer1 in c("L.I","L.II.III", "L.IV","L.V.VI","PROG")){
for(layer2 in c("L.I","L.II.IV","L.V.VI","PROG")){
tt1 = data.frame("genes"= rownames(pca_1[pca_1$hclust %in% groups[[layer1]],])[rownames(pca_1[pca_1$hclust
%in% groups[[layer1]],]) %in% Markers_Loo[[layer2]]])
if (dim(tt1)[1] > 0) {
tt1 = cbind(tt1, "COTAN"=layer1, "Loo."=layer2)
}
tt2 = data.frame("genes"=
rownames(pca_1[!pca_1$hclust %in% unlist(groups),])[rownames(pca_1[!pca_1$hclust %in% unlist(groups),]) %in% Markers_Loo[[layer2]]])
if (dim(tt2)[1] > 0) {
tt2 = cbind(tt2, "COTAN"= "Not Grouped", "Loo."=layer2)
}
specific.genes.table = rbind(specific.genes.table,tt1,tt2)
}
}
specific.genes.table[!(duplicated(specific.genes.table)) , ]
#> genes COTAN Loo.
#> 1 Ndnf L.I L.I
#> 2 Reln L.I L.I
#> 3 Ebf3 L.I L.I
#> 4 Trp73 L.I L.I
#> 5 Lhx1 L.I L.I
#> 6 Lhx5 L.I L.I
#> 7 Sla Not Grouped L.V.VI
#> 8 Pax6 Not Grouped PROG
#> 9 Dkk3 Not Grouped PROG
#> 10 9130024F11Rik L.II.III L.II.IV
#> 11 Satb2 L.II.III L.II.IV
#> 12 Pou3f1 L.II.III L.II.IV
#> 13 Dok5 L.II.III L.II.IV
#> 14 Plxna4 L.II.III L.V.VI
#> 19 Htra1 L.IV PROG
#> 22 Sybu L.V.VI L.V.VI
#> 23 Nfe2l3 L.V.VI L.V.VI
#> 24 Hs3st4 L.V.VI L.V.VI
#> 25 Bcl11b L.V.VI L.V.VI
#> 26 Mc4r L.V.VI L.V.VI
#> 27 Crym L.V.VI L.V.VI
#> 28 Tbr1 L.V.VI L.V.VI
#> 29 Nxph3 L.V.VI L.V.VI
#> 30 Fezf2 L.V.VI L.V.VI
#> 35 Aldoc PROG PROG
#> 36 Ednrb PROG PROG
#> 37 Hes1 PROG PROG
#> 38 Hes5 PROG PROG
#> 39 Gas1 PROG PROG
#> 40 Cyr61 PROG PROG
#> 41 Tk1 PROG PROG
#> 42 Nde1 PROG PROG
#> 43 Tcf19 PROG PROGdend %>%
set("labels", ifelse(labels(dend) %in% rownames(pca_1)[rownames(pca_1) %in% specific.genes.table$genes], labels(dend), "")) %>%
set("branches_k_color", value = c("gray80","#4DBBD5FF","#91D1C2FF" ,"gray80","#F39B7FFF","#E64B35FF","#3C5488FF"), k = 7) %>%
plot(horiz=T, axes=T,xlim = c(0,80),cex = 10, dLeaf = -7)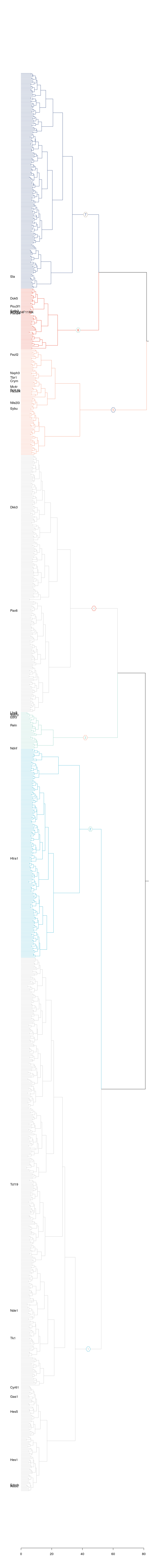
2 WGCNA
library(WGCNA)
#> Loading required package: dynamicTreeCut
#> Loading required package: fastcluster
#>
#> Attaching package: 'fastcluster'
#> The following object is masked from 'package:stats':
#>
#> hclust
#>
#>
#> Attaching package: 'WGCNA'
#> The following object is masked from 'package:stats':
#>
#> cor
library(cluster)
library(data.table)
library(Matrix)
library(Seurat)
#> Attaching SeuratObject
library(utils)
library(dplyr)
#>
#> Attaching package: 'dplyr'
#> The following objects are masked from 'package:data.table':
#>
#> between, first, last
#> The following objects are masked from 'package:stats':
#>
#> filter, lag
#> The following objects are masked from 'package:base':
#>
#> intersect, setdiff, setequal, union
library(patchwork)
library(graphics)
options(stringsAsFactors = FALSE)
data_dir = "Data/"
library(gplots)
#>
#> Attaching package: 'gplots'
#> The following object is masked from 'package:stats':
#>
#> lowess
myheatcol = colorpanel(250,'red',"orange",'lemonchiffon')2.1 Test with the 2000 most varied genes
This seems the best option for the analysis with WGCNA.
data = as.data.frame(fread(paste(input_dir,"E175_only_cortical_cells.txt.gz", sep = "/"),sep = "\t"))
data = as.data.frame(data)
rownames(data) = data$V1
data = data[,2:ncol(data)]
data[1:10,1:10]
#> CGTTTAGTTTAC TCTAGAACAACG ACCTTTGTTCGT TTGTCTTCTTCG TAAAATATCGCC
#> 0610005C13Rik 0 0 0 0 0
#> 0610007N19Rik 0 0 0 0 0
#> 0610007P14Rik 2 1 1 1 0
#> 0610009B22Rik 1 1 0 0 0
#> 0610009D07Rik 2 3 0 3 0
#> 0610009E02Rik 0 0 0 0 0
#> 0610009L18Rik 0 0 0 2 0
#> 0610009O20Rik 0 0 0 0 0
#> 0610010F05Rik 1 2 2 0 1
#> 0610010K14Rik 0 0 0 0 0
#> GTACCCTATTTC GCACATTACCCA CCTCGCGCGGCT TTAATTTTGCCT GTCTTGCGTTTT
#> 0610005C13Rik 0 0 0 0 0
#> 0610007N19Rik 0 0 2 0 0
#> 0610007P14Rik 1 0 0 0 0
#> 0610009B22Rik 0 0 1 1 0
#> 0610009D07Rik 2 1 2 1 5
#> 0610009E02Rik 0 0 0 0 0
#> 0610009L18Rik 0 1 1 2 0
#> 0610009O20Rik 0 0 0 1 0
#> 0610010F05Rik 4 0 1 0 2
#> 0610010K14Rik 0 0 0 0 0E17 <- CreateSeuratObject(counts = data, project = "Cortex E17.5", min.cells = 3, min.features = 200)
#> Warning: Feature names cannot have underscores ('_'), replacing with dashes
#> ('-')
E17[["percent.mt"]] <- PercentageFeatureSet(E17, pattern = "^mt-")
VlnPlot(E17, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)

plot1 <- FeatureScatter(E17, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(E17, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2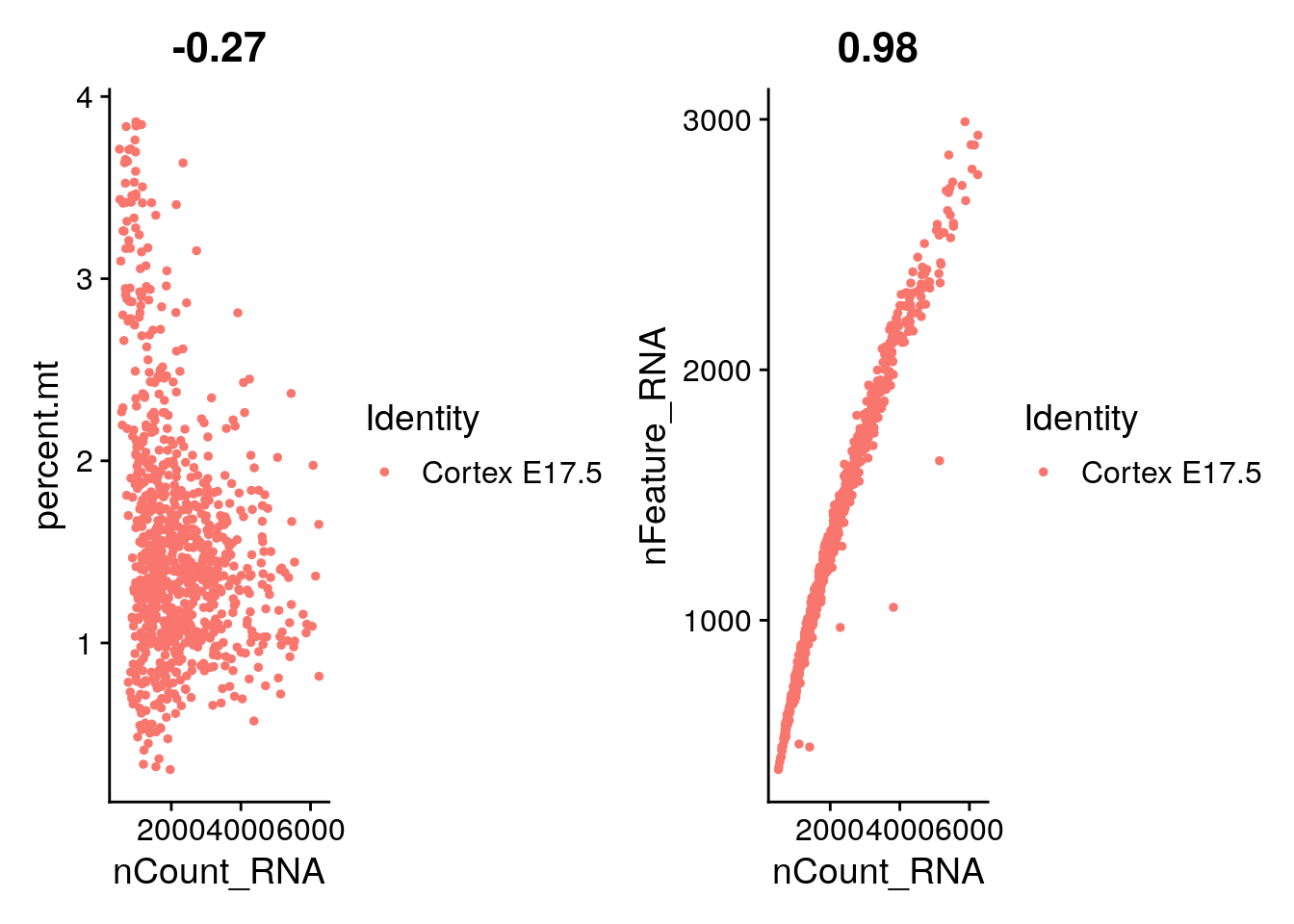
E17 <- NormalizeData(E17, normalization.method = "LogNormalize", scale.factor = 10000)
E17 <- FindVariableFeatures(E17, selection.method = "vst", nfeatures = 2000)
# Identify the 10 most highly variable genes
top10 <- head(VariableFeatures(E17), 10)
# plot variable features with and without labels
plot1 <- VariableFeaturePlot(E17)
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE)
#> When using repel, set xnudge and ynudge to 0 for optimal results
plot2
all.genes <- rownames(E17)
E17 <- ScaleData(E17, features = all.genes)
#> Centering and scaling data matrix
E17 <- RunPCA(E17, features = VariableFeatures(object = E17))
#> PC_ 1
#> Positive: Fabp7, Aldoc, Mfge8, Dbi, Ednrb, Vim, Slc1a3, Mt3, Apoe, Ttyh1
#> Tnc, Sox2, Atp1a2, Ddah1, Hes5, Sparc, Mlc1, Ppap2b, Rgcc, Bcan
#> Ndrg2, Qk, Lxn, Id3, Phgdh, Slc9a3r1, Nr2e1, Aldh1l1, Gpx8, Mt1
#> Negative: Tubb3, Stmn2, Neurod6, Stmn4, Map1b, Stmn1, Myt1l, Mef2c, Thra, 4930506M07Rik
#> Bcl11a, Gap43, Bhlhe22, Syt4, Cntn2, Nell2, Hs6st2, 9130024F11Rik, Olfm1, Satb2
#> Akap9, Ptprd, Rbfox1, Clmp, Ina, Enc1, Camk2b, Dync1i1, Dab1, Atp2b1
#> PC_ 2
#> Positive: Sstr2, Mdk, Meis2, Pou3f2, Eomes, Zbtb20, Unc5d, Sema3c, Fos, Tead2
#> Palmd, Mfap4, Nhlh1, Ulk4, H1f0, Uaca, Neurog2, Neurod1, Ezr, Ier2
#> Nrn1, Baz2b, Pdzrn3, Btg2, Egr1, Mfap2, Loxl1, H2afv, Hbp1, Nnat
#> Negative: Gap43, Sybu, Dync1i1, Meg3, Mef2c, Map1b, Fezf2, Camk2b, Ina, Stmn2
#> Cdh13, Thra, Nin, Rac3, Igfbp3, Ssbp2, Neto2, Cd200, Hmgcs1, Tuba1b
#> Syt1, Slc6a15, Mapre2, Plk2, Rprm, Atp1b1, Cadm2, Arpp21, Kitl, Ntrk2
#> PC_ 3
#> Positive: Meg3, Smpdl3a, Slc9a3r1, Slc15a2, Timp3, Tmem47, Ndrg2, Apoe, Ttyh1, Fmo1
#> Mlc1, Scrg1, Islr2, Malat1, Gstm1, Gja1, Ndnf, Aldh1l1, Mt3, Sparc
#> Serpinh1, Paqr7, Asrgl1, Sepp1, S100a1, Atp1b1, Ctsl, Cpe, S100a16, Lhx5
#> Negative: Birc5, Top2a, Cenpm, Pbk, Tpx2, Cenpe, Mki67, Cdca8, Gmnn, Cks2
#> Ccnb1, Ccnb2, Spc24, Hmgb2, Cenpf, Tk1, Hmmr, Prc1, Kif11, Ccna2
#> 2810417H13Rik, C330027C09Rik, Cdca2, Ect2, Nusap1, Cenpa, Uhrf1, Plk1, Spc25, Knstrn
#> PC_ 4
#> Positive: Lhx5, Nhlh2, Snhg11, Reln, 1500016L03Rik, Trp73, Cacna2d2, Ndnf, Car10, Lhx1
#> Islr2, Pcp4, Meg3, RP24-351J24.2, Rcan2, Pnoc, Mab21l1, Zic1, E330013P04Rik, Emx2
#> Malat1, Ebf3, Nr2f2, Zcchc12, Zbtb20, Celf4, Tmem163, Ache, Calb2, Unc5b
#> Negative: Ptn, Satb2, 9130024F11Rik, Neurod6, Mef2c, Dab1, Limch1, Hs6st2, Abracl, Dok5
#> Gucy1a3, Nell2, Ptprz1, Syt4, Ttc28, Clmp, Macrod2, Fam19a2, Smpdl3a, Ndrg1
#> Gstm1, 4930506M07Rik, Paqr7, Aldh1l1, Myt1l, Hmgcs1, Slc15a2, Pdzrn4, Slc9a3r1, Aldoc
#> PC_ 5
#> Positive: Fam210b, Sfrp1, Pax6, Enkur, Tubb3, Tuba1b, Mcm3, Veph1, Stmn1, Eif1b
#> Map1b, Hopx, Abracl, Cdk2ap2, Tfap2c, Rps27l, 2810025M15Rik, Slc14a2, Prdx1, Hells
#> Gap43, Sept11, Egln3, Gm1840, Ezr, Cpne2, 9130024F11Rik, Nes, Efnb2, Cux1
#> Negative: Serpine2, Id1, Olig1, Sparcl1, Igfbp3, Fam212b, Ccnb2, Ppic, Gng12, Ccnb1
#> Bcan, Cenpe, Pbk, Id3, Rasl11a, Plk1, Aqp4, Aspm, Hmmr, Slc6a1
#> Slc4a4, Malat1, Myo6, Timp3, Meg3, Cdk1, Prrx1, Npy, B2m, Cspg4
DimPlot(E17, reduction = "pca")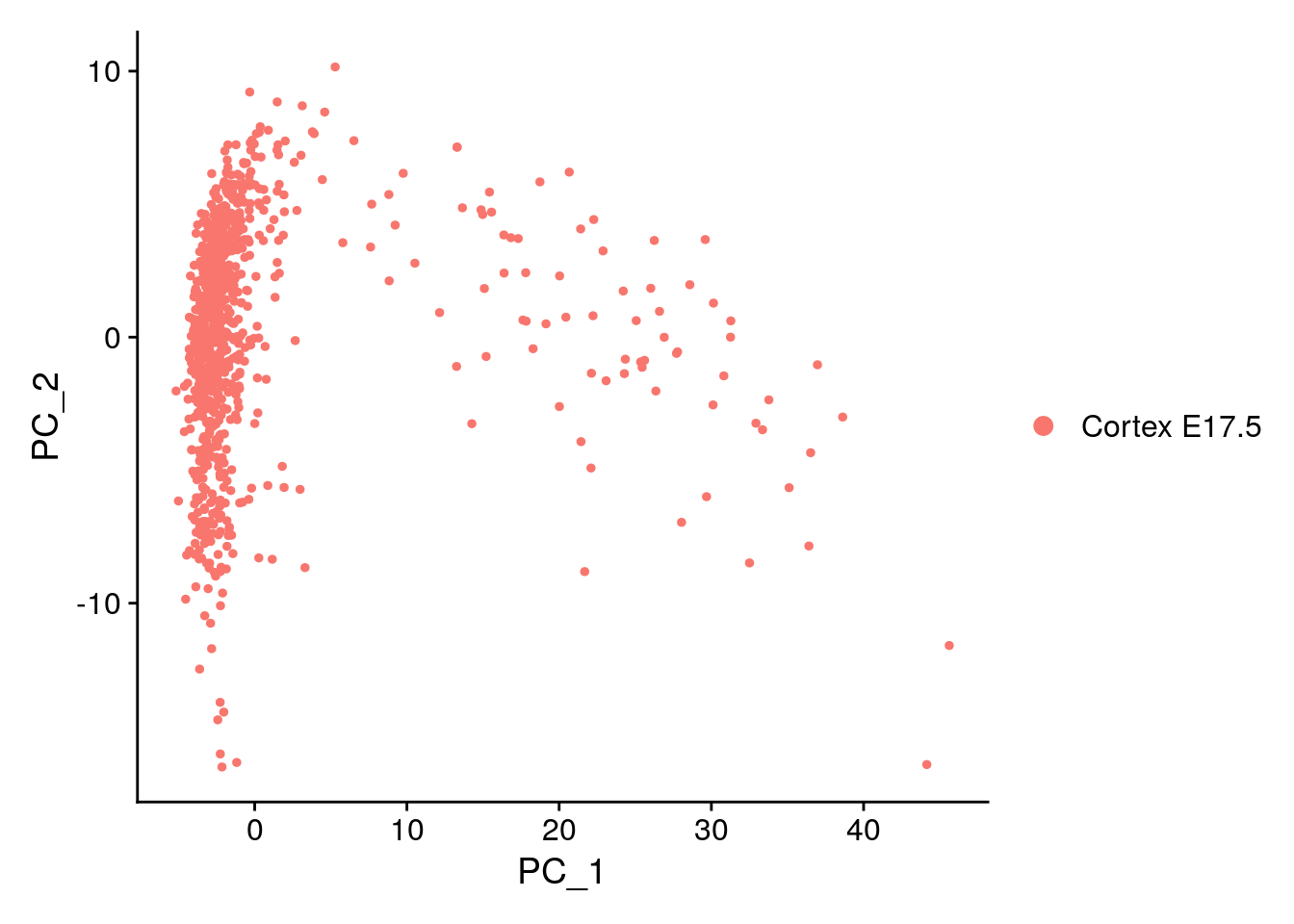
all.genes <- rownames(E17)
E17 <- ScaleData(E17, features = all.genes)
#> Centering and scaling data matrix
seurat.data = as.matrix(E17[["RNA"]]@data)
Var.genes = VariableFeatures(E17)[1:2000]datExpr0 = t(seurat.data[rownames(seurat.data) %in% Var.genes,])
gsg = goodSamplesGenes(datExpr0, verbose = 3)
#> Flagging genes and samples with too many missing values...
#> ..step 1
gsg$allOK
#> [1] TRUE
if (!gsg$allOK){
# Optionally, print the gene and sample names that were removed:
if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")));
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")));
# Remove the offending genes and samples from the data:
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
sampleTree = hclust(dist(datExpr0), method = "average");
# Plot the sample tree: Open a graphic output window of size 12 by 9 inches
# The user should change the dimensions if the window is too large or too small.
sizeGrWindow(12,9)
par(cex = 0.6);
par(mar = c(0,4,2,0))
plot(sampleTree, main = "Sample clustering to detect outliers", sub="", xlab="", cex.lab = 1.5,
cex.axis = 1.5, cex.main = 2)
# Plot a line to show the cut
#abline(h = 400, col = "red")No outliner detected
# Automatic network construction and module detection
# Choose a set of soft-thresholding powers
powers = c(c(1:10), seq(from = 10, to=25, by=2))
# Call the network topology analysis function
sft = pickSoftThreshold(datExpr0, powerVector = powers, verbose = 5)
#> pickSoftThreshold: will use block size 2000.
#> pickSoftThreshold: calculating connectivity for given powers...
#> ..working on genes 1 through 2000 of 2000
#> Warning: executing %dopar% sequentially: no parallel backend registered
#> Power SFT.R.sq slope truncated.R.sq mean.k. median.k. max.k.
#> 1 1 0.882 -3.43 0.9270 6.29e+01 5.70e+01 188.0000
#> 2 2 0.961 -2.50 0.9600 4.90e+00 3.12e+00 41.8000
#> 3 3 0.961 -1.95 0.9610 7.87e-01 2.95e-01 14.4000
#> 4 4 0.927 -1.76 0.9140 2.04e-01 4.01e-02 6.2800
#> 5 5 0.361 -2.33 0.2680 7.08e-02 6.98e-03 3.1400
#> 6 6 0.368 -2.21 0.2850 2.95e-02 1.40e-03 1.7100
#> 7 7 0.345 -2.61 0.2930 1.39e-02 2.82e-04 0.9890
#> 8 8 0.342 -2.47 0.3070 7.12e-03 6.31e-05 0.5930
#> 9 9 0.366 -1.89 0.3020 3.91e-03 1.49e-05 0.3660
#> 10 10 0.311 -1.68 0.1190 2.27e-03 3.43e-06 0.2310
#> 11 10 0.311 -1.68 0.1190 2.27e-03 3.43e-06 0.2310
#> 12 12 0.280 -1.93 0.0743 8.63e-04 2.01e-07 0.1270
#> 13 14 0.379 -2.36 0.3690 3.74e-04 1.25e-08 0.0821
#> 14 16 0.436 -2.70 0.3160 1.79e-04 7.91e-10 0.0545
#> 15 18 0.433 -2.58 0.3590 9.31e-05 5.07e-11 0.0367
#> 16 20 0.468 -2.57 0.3160 5.17e-05 3.32e-12 0.0249
#> 17 22 0.490 -2.45 0.4020 3.02e-05 2.16e-13 0.0170
#> 18 24 0.498 -2.36 0.4110 1.84e-05 1.47e-14 0.0117Plot the results:
sizeGrWindow(9, 5)
par(mfrow = c(1,2));
cex1 = 0.9;
# Scale-free topology fit index as a function of the soft-thresholding power
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n",
main = paste("Scale independence"));
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red");
# this line corresponds to using an R^2 cut-off of h
abline(h=0.90,col="red")
# Mean connectivity as a function of the soft-thresholding power
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",
main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")Tested with 5, 3 and 2 and 4. The best seems 2
net = blockwiseModules(datExpr0, power = 2, maxBlockSize = 20000,
TOMType = "signed", minModuleSize = 30,
reassignThreshold = 0, mergeCutHeight = 0.25,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase = "E17.5",
verbose = 3)
#> Calculating module eigengenes block-wise from all genes
#> Flagging genes and samples with too many missing values...
#> ..step 1
#> ..Working on block 1 .
#> TOM calculation: adjacency..
#> ..will not use multithreading.
#> Fraction of slow calculations: 0.000000
#> ..connectivity..
#> ..matrix multiplication (system BLAS)..
#> ..normalization..
#> ..done.
#> ..saving TOM for block 1 into file E17.5-block.1.RData
#> ....clustering..
#> ....detecting modules..
#> ....calculating module eigengenes..
#> ....checking kME in modules..
#> ..removing 552 genes from module 1 because their KME is too low.
#> ..removing 154 genes from module 2 because their KME is too low.
#> ..removing 86 genes from module 3 because their KME is too low.
#> ..removing 42 genes from module 4 because their KME is too low.
#> ..removing 20 genes from module 5 because their KME is too low.
#> ..merging modules that are too close..
#> mergeCloseModules: Merging modules whose distance is less than 0.25
#> Calculating new MEs...# open a graphics window
sizeGrWindow(12, 9)
# Convert labels to colors for plotting
mergedColors = labels2colors(net$colors)
# Plot the dendrogram and the module colors underneath
plotDendroAndColors(net$dendrograms[[1]], mergedColors[net$blockGenes[[1]]],
"Module colors",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)2.2 Comparition between Loo et al. markers and WGCNA markers
plotNetworkHeatmap(datExpr0, plotGenes = unlist(Markers_Loo),
networkType="signed", useTOM=TRUE,
power=2, main="D. TOM in an signed network")
#> Warning: Not all gene names were recognized. Only the following genes were recognized.
#> Ebf3, Lhx1, Lhx5, Ndnf, Reln, Samd3, Trp73, Satb2, 9130024F11Rik, Dok5, Inhba, Bcl11b, Crym, Fezf2, Hs3st4, Nfe2l3, Nxph3, Rwdd3, Sla, Sybu, Aldoc, Arhgef39, Aspm, Cdc25c, Cyr61, Ednrb, Gas1, Hes1, Hes5, Htra1, Nek2, Pax6, Plk1, Tcf19, Tk1
#> ..connectivity..
#> ..matrix multiplication (system BLAS)..
#> ..normalization..
#> ..done.
plotNetworkHeatmap(datExpr0, plotGenes = unlist(Markers_Loo),
networkType="unsigned", useTOM=TRUE,
power=2, main="D. TOM in an unsigned network")
#> Warning: Not all gene names were recognized. Only the following genes were recognized.
#> Ebf3, Lhx1, Lhx5, Ndnf, Reln, Samd3, Trp73, Satb2, 9130024F11Rik, Dok5, Inhba, Bcl11b, Crym, Fezf2, Hs3st4, Nfe2l3, Nxph3, Rwdd3, Sla, Sybu, Aldoc, Arhgef39, Aspm, Cdc25c, Cyr61, Ednrb, Gas1, Hes1, Hes5, Htra1, Nek2, Pax6, Plk1, Tcf19, Tk1
#> ..connectivity..
#> ..matrix multiplication (system BLAS)..
#> ..normalization..
#> ..done.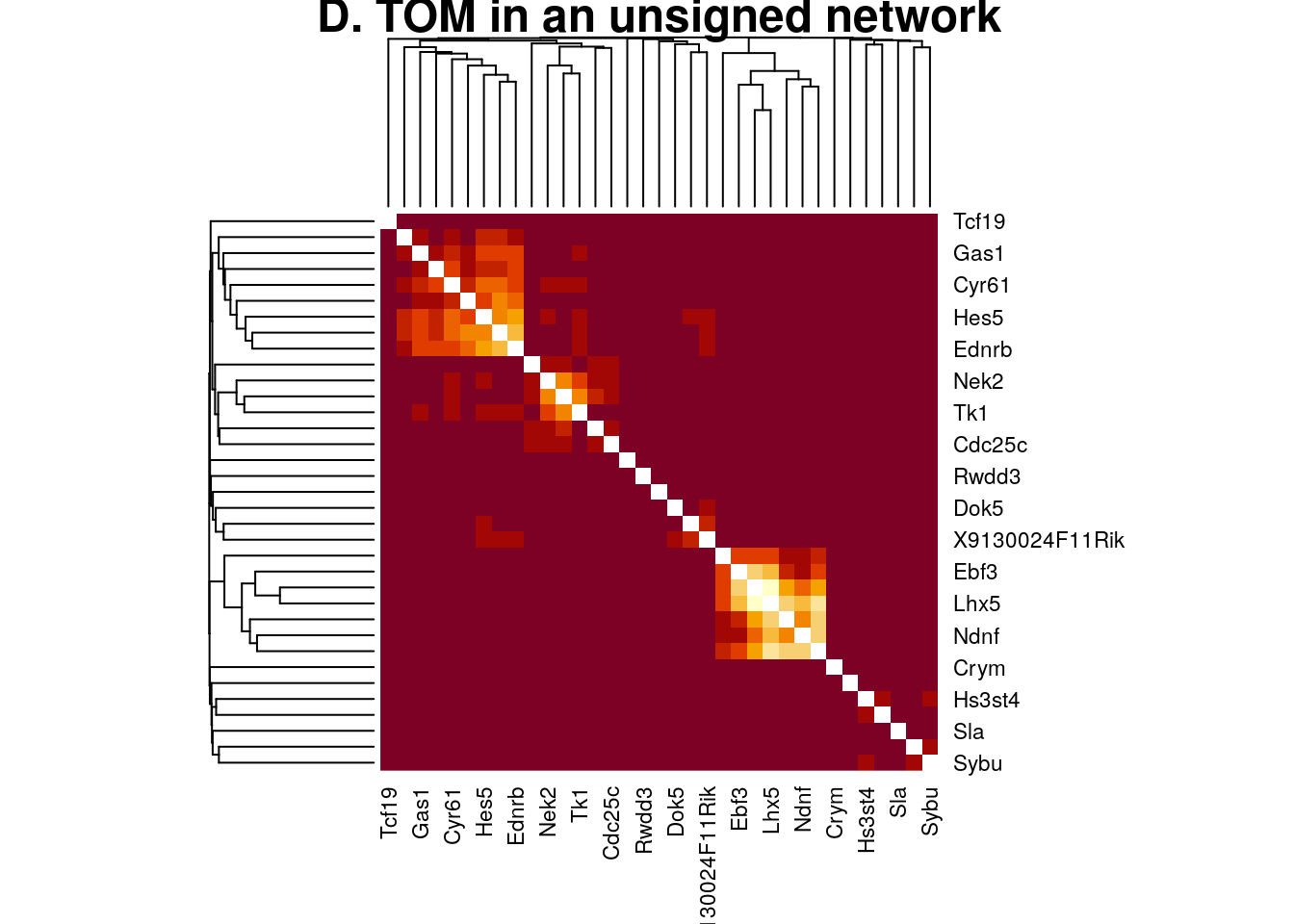
plotNetworkHeatmap(datExpr0, plotGenes = unique(unlist(layers)),
networkType="signed", useTOM=TRUE,
power=2, main="D. TOM in an signed network")
#> ..connectivity..
#> ..matrix multiplication (system BLAS)..
#> ..normalization..
#> ..done.
plotNetworkHeatmap(datExpr0, plotGenes = unique(unlist(layers)),
networkType="unsigned", useTOM=TRUE,
power=2, main="D. TOM in an unsigned network")
#> ..connectivity..
#> ..matrix multiplication (system BLAS)..
#> ..normalization..
#> ..done.
# Calculate topological overlap anew: this could be done more efficiently by saving the TOM
# calculated during module detection, but let us do it again here.
dissTOM = 1-TOMsimilarityFromExpr(datExpr0, power = 2);
#> TOM calculation: adjacency..
#> ..will not use multithreading.
#> Fraction of slow calculations: 0.000000
#> ..connectivity..
#> ..matrix multiplication (system BLAS)..
#> ..normalization..
#> ..done.
# Transform dissTOM with a power to make moderately strong connections more visible in the heatmap
plotTOM = dissTOM^7;
# Set diagonal to NA for a nicer plot
diag(plotTOM) = NA;
rownames(dissTOM)=colnames(datExpr0)
colnames(dissTOM)=colnames(datExpr0)
selectTOM = dissTOM[rownames(dissTOM) %in% unlist(Markers_Loo),colnames(dissTOM) %in% unlist(Markers_Loo)];
# There’s no simple way of restricting a clustering tree to a subset of genes, so we must re-cluster.
selectTree = hclust(as.dist(selectTOM), method = "average")
moduleColors = mergedColors
names(moduleColors) = rownames(dissTOM)
selectColors = moduleColors[names(moduleColors) %in% unlist(Markers_Loo)]
# Open a graphical window
sizeGrWindow(9,9)
# Taking the dissimilarity to a power, say 10, makes the plot more informative by effectively changing
# the color palette; setting the diagonal to NA also improves the clarity of the plot
plotDiss = selectTOM^7;
diag(plotDiss) = NA;
TOMplot(plotDiss, selectTree, selectColors, main = "Network heatmap plot, selected genes",col=myheatcol)plotDendroAndColors(selectTree,selectColors,
"Module colors",
dendroLabels = NULL, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
#> Warning in pmin(objHeights[dendro$order][floor(positions)],
#> objHeights[dendro$order][ceiling(positions)]): an argument will be fractionally
#> recycled
net$colors[names(net$colors) %in% unlist(Markers_Loo)]
#> 9130024F11Rik Aldoc Arhgef39 Aspm Bcl11b
#> 0 1 0 2 0
#> Cdc25c Crym Cyr61 Dok5 Ebf3
#> 2 0 1 0 4
#> Ednrb Fezf2 Gas1 Hes1 Hes5
#> 1 0 1 1 1
#> Hs3st4 Htra1 Inhba Lhx1 Lhx5
#> 0 1 0 4 4
#> Ndnf Nek2 Nfe2l3 Nxph3 Pax6
#> 4 2 0 0 1
#> Plk1 Reln Rwdd3 Samd3 Satb2
#> 2 4 0 0 0
#> Sla Sybu Tcf19 Tk1 Trp73
#> 0 0 0 2 4WGCNA using the 2000 genes most varied by Seurat, detects the following number of markers.
tableMarkersWGCNA = as.data.frame(matrix(nrow = 5,ncol = 4))
colnames(tableMarkersWGCNA)=c("Loo.L.I","Loo.L.II.IV","Loo.L.V.VI","Loo.PROG")
rownames(tableMarkersWGCNA)=c("WGCNA.L.I","WGCNA.Not Grouped","WGCNA.PROG","WGCNA.unknown1", "WGCNA.unknown2")
net$colors[unique(unlist(layers))]
#> Reln Lhx5 Satb2 Cux1 Rorb Sox5 Bcl11b Fezf2 Vim Hes1
#> 4 4 0 0 0 0 0 0 1 1# Attention! The next list need to be updated by hand!
groups = list("L.I"=4,"unknown1"=3,"PROG"=1, "unknown2"=2 , "Not Grouped"=0 )
for(layer1 in c("L.I","unknown1","PROG","unknown2","Not Grouped")){
for(layer2 in c("L.I","L.II.IV", "L.V.VI","PROG")){
tableMarkersWGCNA[paste0("WGCNA.",layer1),paste0("Loo.",layer2)] =
sum(names(net$colors[net$colors %in% groups[[layer1]]]) %in% Markers_Loo[[layer2]])
#tableMarkersWGCNA[paste0("WGCNA.","Not Grouped"),paste0("Loo.",layer2)] =
# sum(names(net$colors[!net$colors %in% unlist(groups)]) %in% Markers_Loo[[layer2]])
}
}
tableMarkersWGCNA
#> Loo.L.I Loo.L.II.IV Loo.L.V.VI Loo.PROG
#> WGCNA.L.I 6 0 0 0
#> WGCNA.Not Grouped 1 4 9 2
#> WGCNA.PROG 0 0 0 8
#> WGCNA.unknown1 0 0 0 0
#> WGCNA.unknown2 0 0 0 5
dend %>%
set("labels", ifelse(labels(dend) %in% rownames(pca_1)[rownames(pca_1) %in% unlist(Markers_Loo)], labels(dend), "")) %>%
set("branches_k_color", value = c("gray80","#4DBBD5FF","#91D1C2FF" ,"gray80","#F39B7FFF","#E64B35FF","#3C5488FF"), k = 7) %>%
set("labels_cex"=0.1) %>%
set("branches_lwd", 0.5) %>%
plot_horiz.dendrogram(horiz=T, axes=T,xlim = c(0,80), dLeaf = -15,text_pos = 3)
sessionInfo()
#> R version 4.0.4 (2021-02-15)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 18.04.5 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/libopenblasp-r0.2.20.so
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] gplots_3.1.1 patchwork_1.1.1 dplyr_1.0.4
#> [4] SeuratObject_4.0.0 Seurat_4.0.1 cluster_2.1.1
#> [7] WGCNA_1.70-3 fastcluster_1.1.25 dynamicTreeCut_1.63-1
#> [10] dendextend_1.14.0 ggrepel_0.9.1 factoextra_1.0.7
#> [13] Matrix_1.3-2 data.table_1.14.0 ggplot2_3.3.3
#> [16] COTAN_0.1.0
#>
#> loaded via a namespace (and not attached):
#> [1] utf8_1.2.1 R.utils_2.10.1 reticulate_1.18
#> [4] tidyselect_1.1.0 RSQLite_2.2.3 AnnotationDbi_1.52.0
#> [7] htmlwidgets_1.5.3 grid_4.0.4 Rtsne_0.15
#> [10] munsell_0.5.0 codetools_0.2-18 ica_1.0-2
#> [13] preprocessCore_1.52.1 future_1.21.0 miniUI_0.1.1.1
#> [16] withr_2.4.1 colorspace_2.0-0 Biobase_2.50.0
#> [19] filelock_1.0.2 highr_0.8 knitr_1.31
#> [22] rstudioapi_0.13 stats4_4.0.4 ROCR_1.0-11
#> [25] tensor_1.5 ggsignif_0.6.1 listenv_0.8.0
#> [28] labeling_0.4.2 polyclip_1.10-0 bit64_4.0.5
#> [31] farver_2.1.0 basilisk_1.2.1 parallelly_1.24.0
#> [34] vctrs_0.3.6 generics_0.1.0 xfun_0.22
#> [37] R6_2.5.0 doParallel_1.0.16 clue_0.3-58
#> [40] bitops_1.0-6 spatstat.utils_2.1-0 cachem_1.0.3
#> [43] assertthat_0.2.1 promises_1.2.0.1 scales_1.1.1
#> [46] nnet_7.3-15 gtable_0.3.0 Cairo_1.5-12.2
#> [49] globals_0.14.0 goftest_1.2-2 rlang_0.4.10
#> [52] GlobalOptions_0.1.2 splines_4.0.4 rstatix_0.7.0
#> [55] lazyeval_0.2.2 impute_1.64.0 spatstat.geom_1.65-5
#> [58] broom_0.7.5 checkmate_2.0.0 yaml_2.2.1
#> [61] reshape2_1.4.4 abind_1.4-5 backports_1.2.1
#> [64] httpuv_1.5.5 Hmisc_4.5-0 tools_4.0.4
#> [67] ellipsis_0.3.1 spatstat.core_1.65-5 jquerylib_0.1.3
#> [70] RColorBrewer_1.1-2 BiocGenerics_0.36.0 ggridges_0.5.3
#> [73] Rcpp_1.0.6 plyr_1.8.6 base64enc_0.1-3
#> [76] purrr_0.3.4 basilisk.utils_1.2.2 deldir_0.2-10
#> [79] ggpubr_0.4.0 rpart_4.1-15 pbapply_1.4-3
#> [82] GetoptLong_1.0.5 viridis_0.5.1 cowplot_1.1.1
#> [85] S4Vectors_0.28.1 zoo_1.8-8 haven_2.3.1
#> [88] magrittr_2.0.1 scattermore_0.7 openxlsx_4.2.3
#> [91] circlize_0.4.12 lmtest_0.9-38 RANN_2.6.1
#> [94] fitdistrplus_1.1-3 matrixStats_0.58.0 hms_1.0.0
#> [97] mime_0.10 evaluate_0.14 xtable_1.8-4
#> [100] rio_0.5.16 jpeg_0.1-8.1 readxl_1.3.1
#> [103] IRanges_2.24.1 gridExtra_2.3 shape_1.4.5
#> [106] compiler_4.0.4 tibble_3.1.0 KernSmooth_2.23-18
#> [109] crayon_1.4.0 R.oo_1.24.0 htmltools_0.5.1.1
#> [112] mgcv_1.8-33 later_1.1.0.1 Formula_1.2-4
#> [115] tidyr_1.1.2 DBI_1.1.1 ComplexHeatmap_2.6.2
#> [118] MASS_7.3-53.1 rappdirs_0.3.3 car_3.0-10
#> [121] R.methodsS3_1.8.1 parallel_4.0.4 igraph_1.2.6
#> [124] forcats_0.5.1 pkgconfig_2.0.3 foreign_0.8-81
#> [127] spatstat.sparse_2.0-0 plotly_4.9.3 foreach_1.5.1
#> [130] bslib_0.2.4 stringr_1.4.0 digest_0.6.27
#> [133] sctransform_0.3.2 RcppAnnoy_0.0.18 spatstat.data_2.1-0
#> [136] rmarkdown_2.7 cellranger_1.1.0 leiden_0.3.7
#> [139] htmlTable_2.1.0 uwot_0.1.10 curl_4.3
#> [142] gtools_3.8.2 shiny_1.6.0 rjson_0.2.20
#> [145] nlme_3.1-152 lifecycle_1.0.0 jsonlite_1.7.2
#> [148] carData_3.0-4 viridisLite_0.3.0 fansi_0.4.2
#> [151] pillar_1.5.1 lattice_0.20-41 fastmap_1.1.0
#> [154] httr_1.4.2 survival_3.2-10 GO.db_3.12.1
#> [157] glue_1.4.2 zip_2.1.1 png_0.1-7
#> [160] iterators_1.0.13 bit_4.0.4 stringi_1.5.3
#> [163] sass_0.3.1 blob_1.2.1 caTools_1.18.1
#> [166] latticeExtra_0.6-29 memoise_2.0.0 irlba_2.3.3
#> [169] future.apply_1.7.0