library (ggplot2)library (tibble)library (zeallot)library (COTAN)library (plyr)library (scales) library (rlang)library (Seurat)library (wordcloud)library (stringr)library (assertr)library (ggVennDiagram)library (ggplot2)library (tidyr)library (enrichR)<- getOption ("enrichR.live" )if (websiteLive) dbs <- listEnrichrDbs ()head (dbs)
geneCoverage genesPerTerm libraryName
1 13362 275 Genome_Browser_PWMs
2 27884 1284 TRANSFAC_and_JASPAR_PWMs
3 6002 77 Transcription_Factor_PPIs
4 47172 1370 ChEA_2013
5 47107 509 Drug_Perturbations_from_GEO_2014
6 21493 3713 ENCODE_TF_ChIP-seq_2014
link numTerms
1 http://hgdownload.cse.ucsc.edu/goldenPath/hg18/database/ 615
2 http://jaspar.genereg.net/html/DOWNLOAD/ 326
3 290
4 http://amp.pharm.mssm.edu/lib/cheadownload.jsp 353
5 http://www.ncbi.nlm.nih.gov/geo/ 701
6 http://genome.ucsc.edu/ENCODE/downloads.html 498
appyter categoryId
1 ea115789fcbf12797fd692cec6df0ab4dbc79c6a 1
2 7d42eb43a64a4e3b20d721fc7148f685b53b6b30 1
3 849f222220618e2599d925b6b51868cf1dab3763 1
4 7ebe772afb55b63b41b79dd8d06ea0fdd9fa2630 7
5 ad270a6876534b7cb063e004289dcd4d3164f342 7
6 497787ebc418d308045efb63b8586f10c526af51 7
#dbs <- "Tabula_Muris" <- "ARCHS4_Tissues" options (parallelly.fork.enable = TRUE )<- "./e15.0_FD_CheckClustersUniformity" setLoggingLevel (1 )setLoggingFile (file.path (outDir, "FindUniformGivenClustersInForebrainDorsal_E150.log" ))
<- readRDS (file = file.path ("Data/MouseCortexFromLoom/SourceData/" , "e15.0_ForebrainDorsal.cotan.RDS" ))<- readRDS (file = file.path ("Data/MouseCortexFromLoom/" , "e15.0_ForebrainDorsal.cotan.RDS" ))
Align to cleaned cells’ list
<- getMetadataCells (fb150ObjRaw)[getCells (fb150Obj), ]#metaCDrop <- getMetadataCells(fb150ObjRaw)[!getCells(fb150ObjRaw) %in% getCells(fb150Obj), ]
Extract the cells of class ‘Neuron’
<- metaC[metaC[["Class" ]] == "Neuron" , ]sort (table (metaNeuron[["Subclass" ]]), decreasing = TRUE )
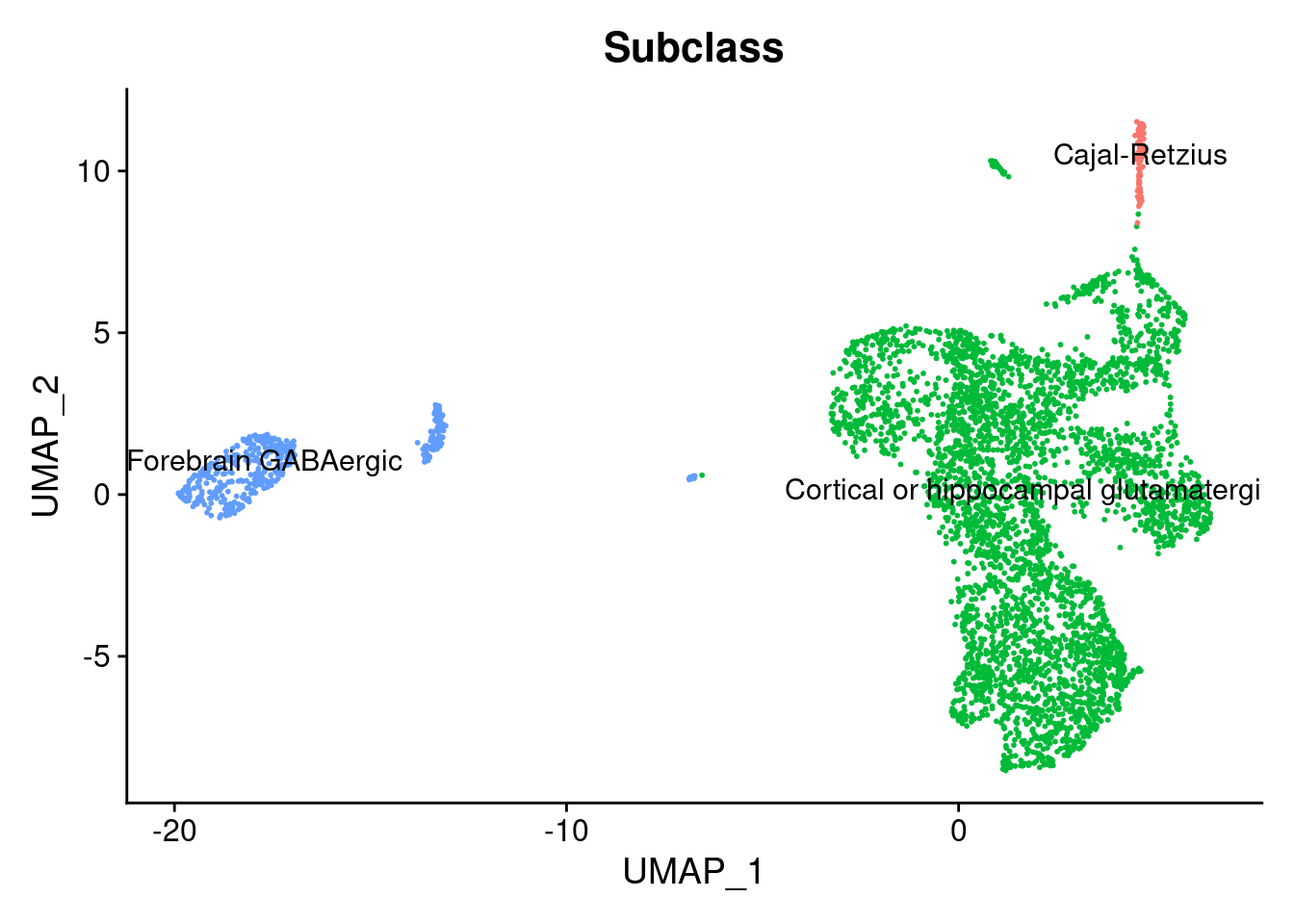
Cortical or hippocampal glutamatergic Forebrain GABAergic
3969 610
Cajal-Retzius Mixed region GABAergic
145 21
Undefined Forebrain glutamatergic
16 15
Hypothalamus Mixed region glutamatergic
8 5
Mixed region and neurotransmitter Hindbrain glutamatergic
4 2
Hindbrain glycinergic Hypothalamus glutamatergic
2 2
Dorsal midbrain glutamatergic Mixed region
1 1
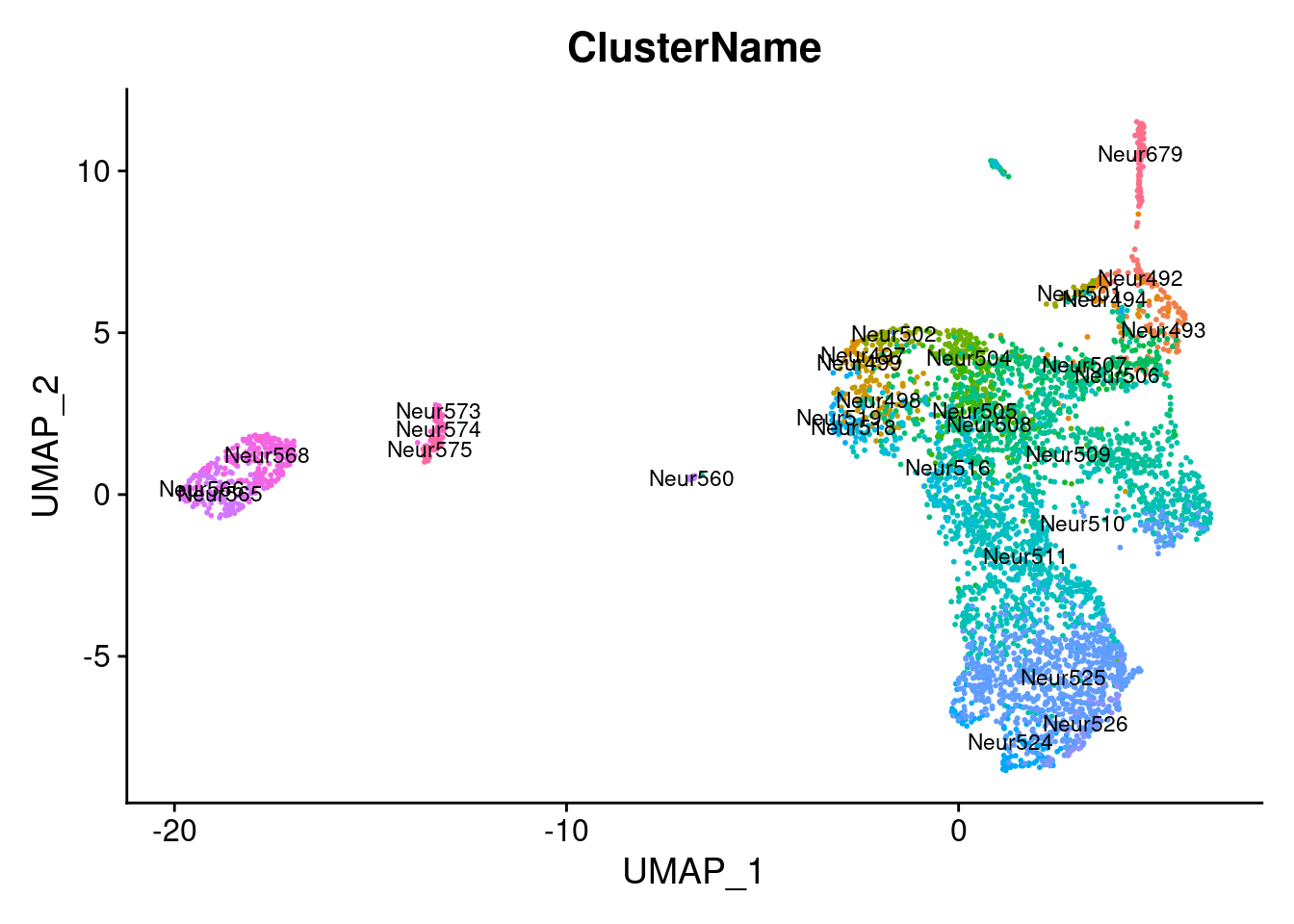
sort (table (metaNeuron[["ClusterName" ]]), decreasing = TRUE )
Neur525 Neur511 Neur509 Neur510 Neur508 Neur507 Neur568 Neur504 Neur505 Neur516
826 540 402 402 397 183 181 174 147 137
Neur565 Neur524 Neur679 Neur493 Neur498 Neur497 Neur506 Neur502 Neur494 Neur574
133 108 105 93 79 51 46 42 41 41
Neur575 Neur492 Neur519 Neur526 Neur566 Neur501 Neur573 Neur499 Neur518 Neur560
41 38 31 28 28 24 24 23 22 20
Neur514 Neur523 Neur569 Neur557 Neur495 Neur520 Neur535 Neur542 Neur677 Neur527
19 19 18 16 15 15 14 14 14 13
Neur496 Neur512 Neur676 Neur517 Neur558 Neur503 Neur739 Neur559 Neur564 Neur538
11 11 11 10 9 8 8 7 7 6
Neur549 Neur561 Neur671 Neur695 Neur738 Neur747 Neur500 Neur536 Neur678 Neur534
6 6 6 6 6 6 5 5 5 4
Neur550 Neur570 Neur686 Neur731 Neur737 Neur513 Neur515 Neur528 Neur533 Neur539
4 4 4 4 4 3 3 3 3 3
Neur544 Neur571 Neur674 Neur675 Neur732 Neur531 Neur543 Neur548 Neur552 Neur554
3 3 3 3 3 2 2 2 2 2
Neur562 Neur670 Neur689 Neur740 Neur529 Neur530 Neur532 Neur537 Neur540 Neur553
2 2 2 2 1 1 1 1 1 1
Neur567 Neur572 Neur601 Neur614 Neur634 Neur647 Neur649 Neur672 Neur680 Neur681
1 1 1 1 1 1 1 1 1 1
Neur684 Neur693 Neur696 Neur726 Neur734 Neur749 Neur750 Neur751 Neur760 Neur771
1 1 1 1 1 1 1 1 1 1
<- names (table (metaNeuron[["ClusterName" ]])[table (metaNeuron[["ClusterName" ]]) >= 20L])<- metaNeuron[metaNeuron$ ClusterName %in% cl.to.keep,]dim (meta.to.keep)
Dropped cell
dim (metaNeuron)[1 ] - dim (meta.to.keep)[1 ]
Removing cells
<- getCells (fb150Obj)[! getCells (fb150Obj) %in% rownames (meta.to.keep)]<- dropGenesCells (fb150Obj,cells = cell.to.drop)dim (meta.to.keep)[1 ] == dim (getRawData (fb150Obj))[2 ]<- getMetadataCells (fb150ObjRaw)[getCells (fb150Obj), ]
<- clean (fb150Obj)<- estimateDispersionBisection (fb150Obj)identical (rownames (fb150Obj@ metaCells), rownames (metaC))@ metaCells <- cbind (fb150Obj@ metaCells,metaC)@ metaCells[1 : 10 ,]
feCells nu feCells nu Age
10X74_4_A_1:TGGTAGACCCTACCx FALSE 0.9375770 FALSE 0.9214807 e15.0
10X73_3_A_1:TACCGGCTTCAGTGx FALSE 0.7855991 FALSE 0.7722440 e15.0
10X74_4_A_1:CCCAGTTGGAGGTGx FALSE 0.6983103 FALSE 0.6863260 e15.0
10X73_3_A_1:AATTGATGAGAATGx FALSE 1.1751850 FALSE 1.1548031 e15.0
10X74_4_A_1:TAGTGGTGAGTAGAx FALSE 1.1001290 FALSE 1.0808973 e15.0
10X74_4_A_1:ATTATGGACTACGAx FALSE 1.3074658 FALSE 1.2844945 e15.0
10X64_3_A_1:TGTATCTGCCTTATx FALSE 1.4138296 FALSE 1.3891434 e15.0
10X64_3_A_1:TAGTATGACTATGGx FALSE 1.6528889 FALSE 1.6249089 e15.0
10X73_3_A_1:ACTTGGGATTGCAGx FALSE 0.7482785 FALSE 0.7355965 e15.0
10X74_4_A_1:AGCTCGCTCCATGAx FALSE 1.1109106 FALSE 1.0914844 e15.0
CellCycle CellID
10X74_4_A_1:TGGTAGACCCTACCx 0.00308982564555286 10X74_4_A_1:TGGTAGACCCTACCx
10X73_3_A_1:TACCGGCTTCAGTGx 0.000789058390320884 10X73_3_A_1:TACCGGCTTCAGTGx
10X74_4_A_1:CCCAGTTGGAGGTGx 0.00177988727380599 10X74_4_A_1:CCCAGTTGGAGGTGx
10X73_3_A_1:AATTGATGAGAATGx 0.000880436696601514 10X73_3_A_1:AATTGATGAGAATGx
10X74_4_A_1:TAGTGGTGAGTAGAx 0.000752021056589585 10X74_4_A_1:TAGTGGTGAGTAGAx
10X74_4_A_1:ATTATGGACTACGAx 0.00174133291119202 10X74_4_A_1:ATTATGGACTACGAx
10X64_3_A_1:TGTATCTGCCTTATx 0.000878348704435661 10X64_3_A_1:TGTATCTGCCTTATx
10X64_3_A_1:TAGTATGACTATGGx 0.00125156445556946 10X64_3_A_1:TAGTATGACTATGGx
10X73_3_A_1:ACTTGGGATTGCAGx 0.00221177771633951 10X73_3_A_1:ACTTGGGATTGCAGx
10X74_4_A_1:AGCTCGCTCCATGAx 0.00111835973904939 10X74_4_A_1:AGCTCGCTCCATGAx
Cell_Conc Chemistry ChipID Class ClusterName
10X74_4_A_1:TGGTAGACCCTACCx 600 v2 10X74 Neuron Neur492
10X73_3_A_1:TACCGGCTTCAGTGx 600 v2 10X73 Neuron Neur492
10X74_4_A_1:CCCAGTTGGAGGTGx 600 v2 10X74 Neuron Neur492
10X73_3_A_1:AATTGATGAGAATGx 600 v2 10X73 Neuron Neur492
10X74_4_A_1:TAGTGGTGAGTAGAx 600 v2 10X74 Neuron Neur492
10X74_4_A_1:ATTATGGACTACGAx 600 v2 10X74 Neuron Neur492
10X64_3_A_1:TGTATCTGCCTTATx 600 v2 10X64 Neuron Neur492
10X64_3_A_1:TAGTATGACTATGGx 600 v2 10X64 Neuron Neur492
10X73_3_A_1:ACTTGGGATTGCAGx 600 v2 10X73 Neuron Neur492
10X74_4_A_1:AGCTCGCTCCATGAx 600 v2 10X74 Neuron Neur492
Clusters Date_Captured DonorID
10X74_4_A_1:TGGTAGACCCTACCx 492 2016-11-30 Batch14K-2
10X73_3_A_1:TACCGGCTTCAGTGx 492 2016-11-30 Batch14K-2
10X74_4_A_1:CCCAGTTGGAGGTGx 492 2016-11-30 Batch14K-2
10X73_3_A_1:AATTGATGAGAATGx 492 2016-11-30 Batch14K-2
10X74_4_A_1:TAGTGGTGAGTAGAx 492 2016-11-30 Batch14K-2
10X74_4_A_1:ATTATGGACTACGAx 492 2016-11-30 Batch14K-2
10X64_3_A_1:TGTATCTGCCTTATx 492 2016-11-15 Batch11H-5
10X64_3_A_1:TAGTATGACTATGGx 492 2016-11-15 Batch11H-5
10X73_3_A_1:ACTTGGGATTGCAGx 492 2016-11-30 Batch14K-2
10X74_4_A_1:AGCTCGCTCCATGAx 492 2016-11-30 Batch14K-2
DoubletFinderPCA HPF_LogPP IsCycling
10X74_4_A_1:TGGTAGACCCTACCx 0 -881923.697556259 0
10X73_3_A_1:TACCGGCTTCAGTGx 0 -621646.228431766 0
10X74_4_A_1:CCCAGTTGGAGGTGx 0.0303030303030303 -615788.630071891 0
10X73_3_A_1:AATTGATGAGAATGx 0.027027027027027 -1043580.43446046 0
10X74_4_A_1:TAGTGGTGAGTAGAx 0.0606060606060606 -892588.390822332 0
10X74_4_A_1:ATTATGGACTACGAx 0.0606060606060606 -1057464.46525045 0
10X64_3_A_1:TGTATCTGCCTTATx 0.0588235294117647 -1259381.55581998 0
10X64_3_A_1:TAGTATGACTATGGx 0.147058823529412 -1384562.64613975 0
10X73_3_A_1:ACTTGGGATTGCAGx 0.027027027027027 -766332.582748427 0
10X74_4_A_1:AGCTCGCTCCATGAx 0 -760899.629760664 0
Label Location_E9_E11 NCellsCluster NGenes
10X74_4_A_1:TGGTAGACCCTACCx G119 nan 457 2402
10X73_3_A_1:TACCGGCTTCAGTGx G115 nan 457 2034
10X74_4_A_1:CCCAGTTGGAGGTGx G119 nan 457 1915
10X73_3_A_1:AATTGATGAGAATGx G115 nan 457 2555
10X74_4_A_1:TAGTGGTGAGTAGAx G119 nan 457 2448
10X74_4_A_1:ATTATGGACTACGAx G119 nan 457 2785
10X64_3_A_1:TGTATCTGCCTTATx G94 nan 457 2842
10X64_3_A_1:TAGTATGACTATGGx G94 nan 457 3160
10X73_3_A_1:ACTTGGGATTGCAGx G115 nan 457 2033
10X74_4_A_1:AGCTCGCTCCATGAx G119 nan 457 2469
Num_Pooled_Animals PCR_Cycles Plug_Date Project
10X74_4_A_1:TGGTAGACCCTACCx 3 13 2016-11-15 Dev
10X73_3_A_1:TACCGGCTTCAGTGx 3 13 2016-11-15 Dev
10X74_4_A_1:CCCAGTTGGAGGTGx 3 13 2016-11-15 Dev
10X73_3_A_1:AATTGATGAGAATGx 3 13 2016-11-15 Dev
10X74_4_A_1:TAGTGGTGAGTAGAx 3 13 2016-11-15 Dev
10X74_4_A_1:ATTATGGACTACGAx 3 13 2016-11-15 Dev
10X64_3_A_1:TGTATCTGCCTTATx 3 13 2016-10-31 Dev
10X64_3_A_1:TAGTATGACTATGGx 3 13 2016-10-31 Dev
10X73_3_A_1:ACTTGGGATTGCAGx 3 13 2016-11-15 Dev
10X74_4_A_1:AGCTCGCTCCATGAx 3 13 2016-11-15 Dev
PseudoAge PseudoTissue Region SampleID
10X74_4_A_1:TGGTAGACCCTACCx 15.905 ForebrainDorsal Forebrain 10X74_4
10X73_3_A_1:TACCGGCTTCAGTGx 13.155 ForebrainDorsal Forebrain 10X73_3
10X74_4_A_1:CCCAGTTGGAGGTGx 13.935 ForebrainDorsal Forebrain 10X74_4
10X73_3_A_1:AATTGATGAGAATGx 15.54 ForebrainDorsal Forebrain 10X73_3
10X74_4_A_1:TAGTGGTGAGTAGAx 14.595 ForebrainDorsal Forebrain 10X74_4
10X74_4_A_1:ATTATGGACTACGAx 14.3725 ForebrainDorsal Forebrain 10X74_4
10X64_3_A_1:TGTATCTGCCTTATx 15.1275 ForebrainDorsal Forebrain 10X64_3
10X64_3_A_1:TAGTATGACTATGGx 16.225 ForebrainDorsal Forebrain 10X64_3
10X73_3_A_1:ACTTGGGATTGCAGx 12.8175 ForebrainVentral Forebrain 10X73_3
10X74_4_A_1:AGCTCGCTCCATGAx 14.7975 ForebrainDorsal Forebrain 10X74_4
SampleName Sample_Index Sex Species Split Strain
10X74_4_A_1:TGGTAGACCCTACCx G119 SI-3A-E7 ? Mm 0 CD-1
10X73_3_A_1:TACCGGCTTCAGTGx G115 SI-3A-A7 ? Mm 0 CD-1
10X74_4_A_1:CCCAGTTGGAGGTGx G119 SI-3A-E7 ? Mm 0 CD-1
10X73_3_A_1:AATTGATGAGAATGx G115 SI-3A-A7 ? Mm 0 CD-1
10X74_4_A_1:TAGTGGTGAGTAGAx G119 SI-3A-E7 ? Mm 0 CD-1
10X74_4_A_1:ATTATGGACTACGAx G119 SI-3A-E7 ? Mm 0 CD-1
10X64_3_A_1:TGTATCTGCCTTATx G94 SI-3A-H3 ? Mm 0 CD-1
10X64_3_A_1:TAGTATGACTATGGx G94 SI-3A-H3 ? Mm 0 CD-1
10X73_3_A_1:ACTTGGGATTGCAGx G115 SI-3A-A7 ? Mm 0 CD-1
10X74_4_A_1:AGCTCGCTCCATGAx G119 SI-3A-E7 ? Mm 0 CD-1
Subclass
10X74_4_A_1:TGGTAGACCCTACCx Cortical or hippocampal glutamatergic
10X73_3_A_1:TACCGGCTTCAGTGx Cortical or hippocampal glutamatergic
10X74_4_A_1:CCCAGTTGGAGGTGx Cortical or hippocampal glutamatergic
10X73_3_A_1:AATTGATGAGAATGx Cortical or hippocampal glutamatergic
10X74_4_A_1:TAGTGGTGAGTAGAx Cortical or hippocampal glutamatergic
10X74_4_A_1:ATTATGGACTACGAx Cortical or hippocampal glutamatergic
10X64_3_A_1:TGTATCTGCCTTATx Cortical or hippocampal glutamatergic
10X64_3_A_1:TAGTATGACTATGGx Cortical or hippocampal glutamatergic
10X73_3_A_1:ACTTGGGATTGCAGx Cortical or hippocampal glutamatergic
10X74_4_A_1:AGCTCGCTCCATGAx Cortical or hippocampal glutamatergic
Target_Num_Cells Tissue TotalUMI
10X74_4_A_1:TGGTAGACCCTACCx 3500 ForebrainDorsal 4531
10X73_3_A_1:TACCGGCTTCAGTGx 3500 ForebrainDorsal 3802
10X74_4_A_1:CCCAGTTGGAGGTGx 3500 ForebrainDorsal 3371
10X73_3_A_1:AATTGATGAGAATGx 3500 ForebrainDorsal 5679
10X74_4_A_1:TAGTGGTGAGTAGAx 3500 ForebrainDorsal 5319
10X74_4_A_1:ATTATGGACTACGAx 3500 ForebrainDorsal 6317
10X64_3_A_1:TGTATCTGCCTTATx 3500 ForebrainDorsal 6831
10X64_3_A_1:TAGTATGACTATGGx 3500 ForebrainDorsal 7990
10X73_3_A_1:ACTTGGGATTGCAGx 3500 ForebrainDorsal 3617
10X74_4_A_1:AGCTCGCTCCATGAx 3500 ForebrainDorsal 5365
Transcriptome cDNA_Lib_Ok ngperul_cDNA
10X74_4_A_1:TGGTAGACCCTACCx mm10 Y 3,1
10X73_3_A_1:TACCGGCTTCAGTGx mm10 Y 3,4
10X74_4_A_1:CCCAGTTGGAGGTGx mm10 Y 3,1
10X73_3_A_1:AATTGATGAGAATGx mm10 Y 3,4
10X74_4_A_1:TAGTGGTGAGTAGAx mm10 Y 3,1
10X74_4_A_1:ATTATGGACTACGAx mm10 Y 3,1
10X64_3_A_1:TGTATCTGCCTTATx mm10 Y 3,5
10X64_3_A_1:TAGTATGACTATGGx mm10 Y 3,5
10X73_3_A_1:ACTTGGGATTGCAGx mm10 Y 3,4
10X74_4_A_1:AGCTCGCTCCATGAx mm10 Y 3,1
Cotan differential expression
DEA on Subclass groups formed by just three cell types: Cortical or hippocampal glutamatergic, Forebrain GABAergic and Cajal-Retzius.
<- set_names (fb150Obj@ metaCells$ Subclass,nm = rownames (fb150Obj@ metaCells))<- DEAOnClusters (fb150Obj,clusters = clusters.cells)head (dea.Subclass$ coex)
Cajal-Retzius Cortical or hippocampal glutamatergic Forebrain GABAergic
Lamc1 -0.026166913 -0.003964787 0.02109719
Lama1 -0.021046008 -0.059311990 0.08266931
Hs3st1 0.009828396 0.072980864 -0.08910190
Fabp3 -0.090809976 0.106706770 -0.06883822
Nrg2 -0.001594555 -0.025294811 0.03037227
Bend4 0.009762501 -0.192484391 0.21811993
DEA on ClusterName: [1] “Neur492” “Neur493” “Neur494” “Neur497” “Neur498” “Neur499” “Neur501” “Neur502” “Neur504” [10] “Neur505” “Neur506” “Neur507” “Neur508” “Neur509” “Neur510” “Neur511” “Neur516” “Neur518” [19] “Neur519” “Neur524” “Neur525” “Neur526” “Neur560” “Neur565” “Neur566” “Neur568” “Neur573” [28] “Neur574” “Neur575” “Neur679”
30 different cell groups.
<- set_names (fb150Obj@ metaCells$ ClusterName,nm = rownames (fb150Obj@ metaCells))<- DEAOnClusters (fb150Obj,clusters = clusters.cells)head (dea.ClusterName$ coex)
Neur492 Neur493 Neur494 Neur497 Neur498
Lamc1 -0.015142482 0.0052112095 -0.007566741 0.026741619 0.010599224
Lama1 -0.009632159 -0.0002343889 -0.010660671 -0.011372217 -0.004549478
Hs3st1 0.017614822 -0.0670123869 0.031852014 -0.033595679 -0.051990356
Fabp3 -0.013876113 0.0978372006 0.033870283 -0.018363993 -0.001097806
Nrg2 -0.005434727 0.0035304650 0.005300480 -0.008163350 -0.009236954
Bend4 -0.006635644 -0.0051243739 -0.007347493 -0.007827941 -0.014149243
Neur499 Neur501 Neur502 Neur504 Neur505
Lamc1 -0.002987865 0.010825673 -0.020364010 -0.01240859 -0.008362820
Lama1 -0.004586508 -0.004627726 0.008897717 -0.02182153 0.013489025
Hs3st1 -0.028752186 0.116297848 0.039591917 0.09582077 0.080567467
Fabp3 -0.001637069 -0.017191649 -0.010664235 -0.06640263 -0.046594627
Nrg2 -0.010451945 0.004869471 0.015748204 -0.02610886 -0.001227308
Bend4 0.027951286 0.012358515 -0.004432487 -0.01802341 -0.003364840
Neur506 Neur507 Neur508 Neur509 Neur510
Lamc1 -0.0137768308 0.013438810 0.003655012 0.015332401 -0.022982461
Lama1 0.0473493936 -0.001843277 -0.020074771 -0.004309732 -0.022499414
Hs3st1 0.0009883989 -0.044753051 -0.005629901 0.113439645 -0.017375581
Fabp3 0.0495867141 -0.030231847 -0.043623601 -0.077834754 0.043983043
Nrg2 -0.0005773858 -0.024882460 -0.051351834 -0.033940039 -0.006889284
Bend4 -0.0096820697 -0.023076428 -0.024799514 -0.026800103 -0.023813556
Neur511 Neur516 Neur518 Neur519 Neur524
Lamc1 -0.006029846 0.029054227 -0.001584281 -0.023778455 0.010622135
Lama1 -0.007894555 -0.022458902 -0.008383886 -0.008774730 -0.008985542
Hs3st1 0.026348751 0.015910202 -0.022958194 -0.011012483 -0.026955014
Fabp3 0.025743620 -0.017541959 0.038232083 0.016478606 0.086814711
Nrg2 -0.041561278 0.008453141 -0.002869805 0.037131271 0.077391246
Bend4 -0.032179960 0.006955173 -0.005798012 -0.006054586 0.010811234
Neur525 Neur526 Neur560 Neur565 Neur566
Lamc1 -0.005698386 8.098411e-03 -0.002777033 0.02760694 0.042467086
Lama1 0.021459376 -5.210111e-03 0.026547559 0.05971741 0.026773138
Hs3st1 -0.058540117 -5.159973e-03 -0.028545565 -0.03421586 -0.018237097
Fabp3 0.104026536 -4.399868e-05 -0.031478717 -0.01143598 -0.007057800
Nrg2 0.067456393 1.888367e-02 0.036151663 0.04125869 0.005918738
Bend4 -0.035214002 -3.580672e-03 -0.003135888 0.21473720 0.012669639
Neur568 Neur573 Neur574 Neur575 Neur679
Lamc1 -0.0238073082 0.016259061 0.029273702 -0.006876455 -0.026166913
Lama1 0.0269694769 0.011948982 -0.005923828 0.009594151 -0.021046008
Hs3st1 -0.0557759330 -0.025415472 -0.033200908 -0.024987623 0.009828396
Fabp3 -0.0650712806 -0.026079990 -0.007401114 -0.012669512 -0.090809976
Nrg2 0.0004859691 -0.008135245 0.001663527 -0.013282661 -0.001594555
Bend4 0.1394771632 -0.002429324 -0.004063790 0.011455264 0.009762501
Seurat object creation
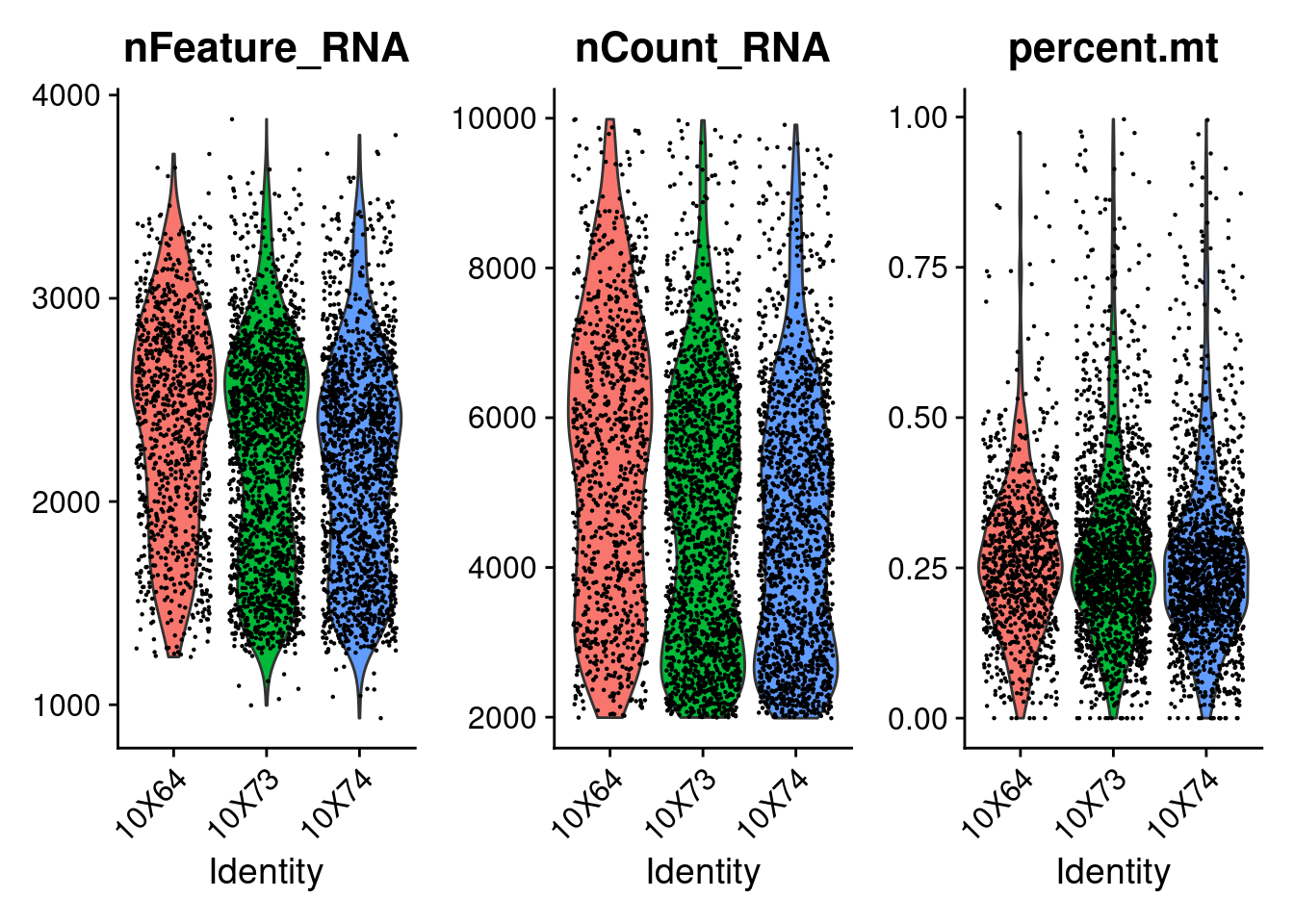
<- CreateSeuratObject (counts = getRawData (fb150Obj), project = "fb15.0" , min.cells = 3 , min.features = 200 )"percent.mt" ]] <- PercentageFeatureSet (seurat.obj, pattern = "^mt." )VlnPlot (seurat.obj, features = c ("nFeature_RNA" , "nCount_RNA" , "percent.mt" ), ncol = 3 )
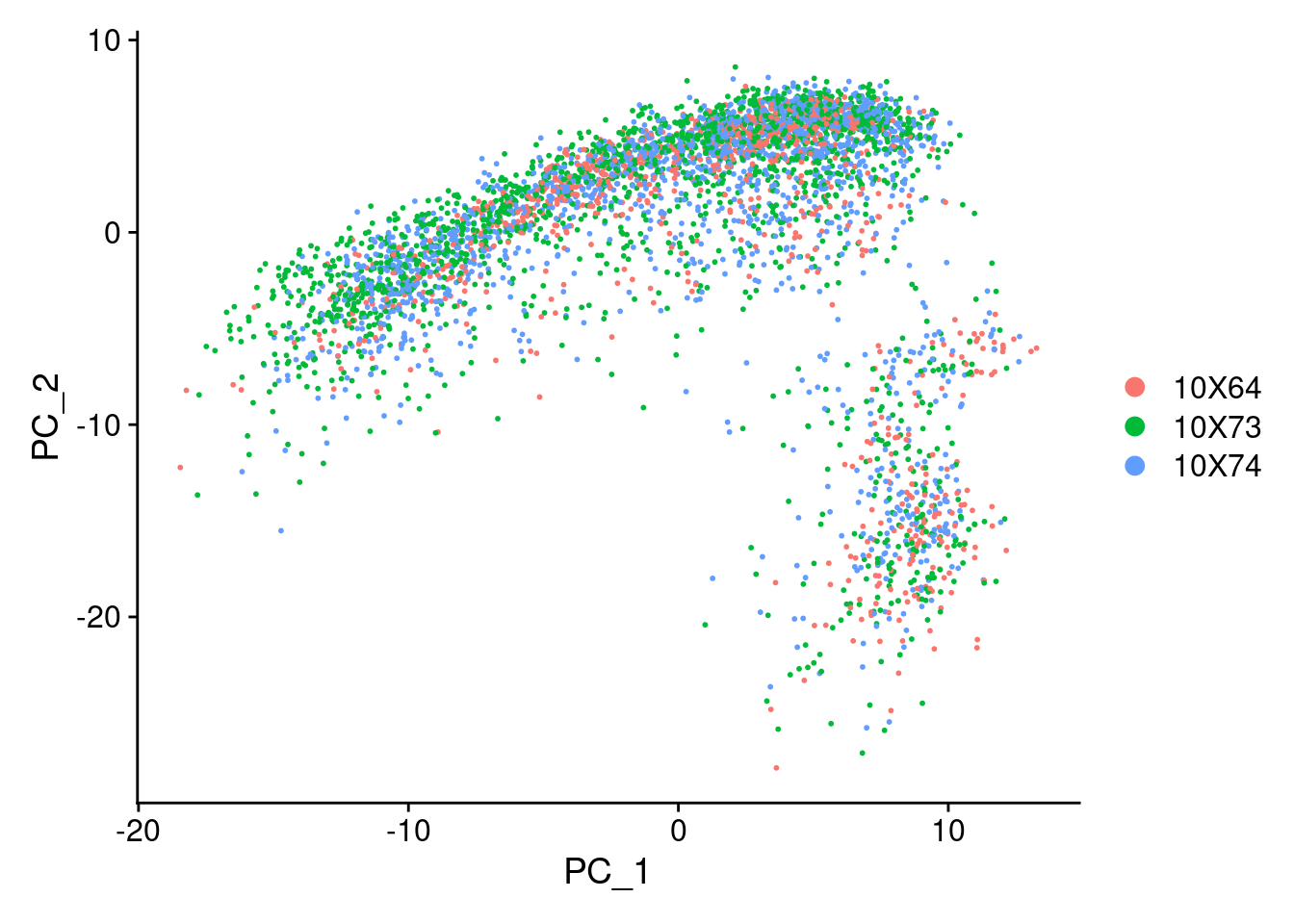
<- NormalizeData (seurat.obj, normalization.method = "LogNormalize" , scale.factor = 10000 )<- FindVariableFeatures (seurat.obj, selection.method = "vst" , nfeatures = 2000 )<- rownames (seurat.obj)<- ScaleData (seurat.obj, features = all.genes)<- RunPCA (seurat.obj, features = VariableFeatures (object = seurat.obj))DimPlot (seurat.obj)
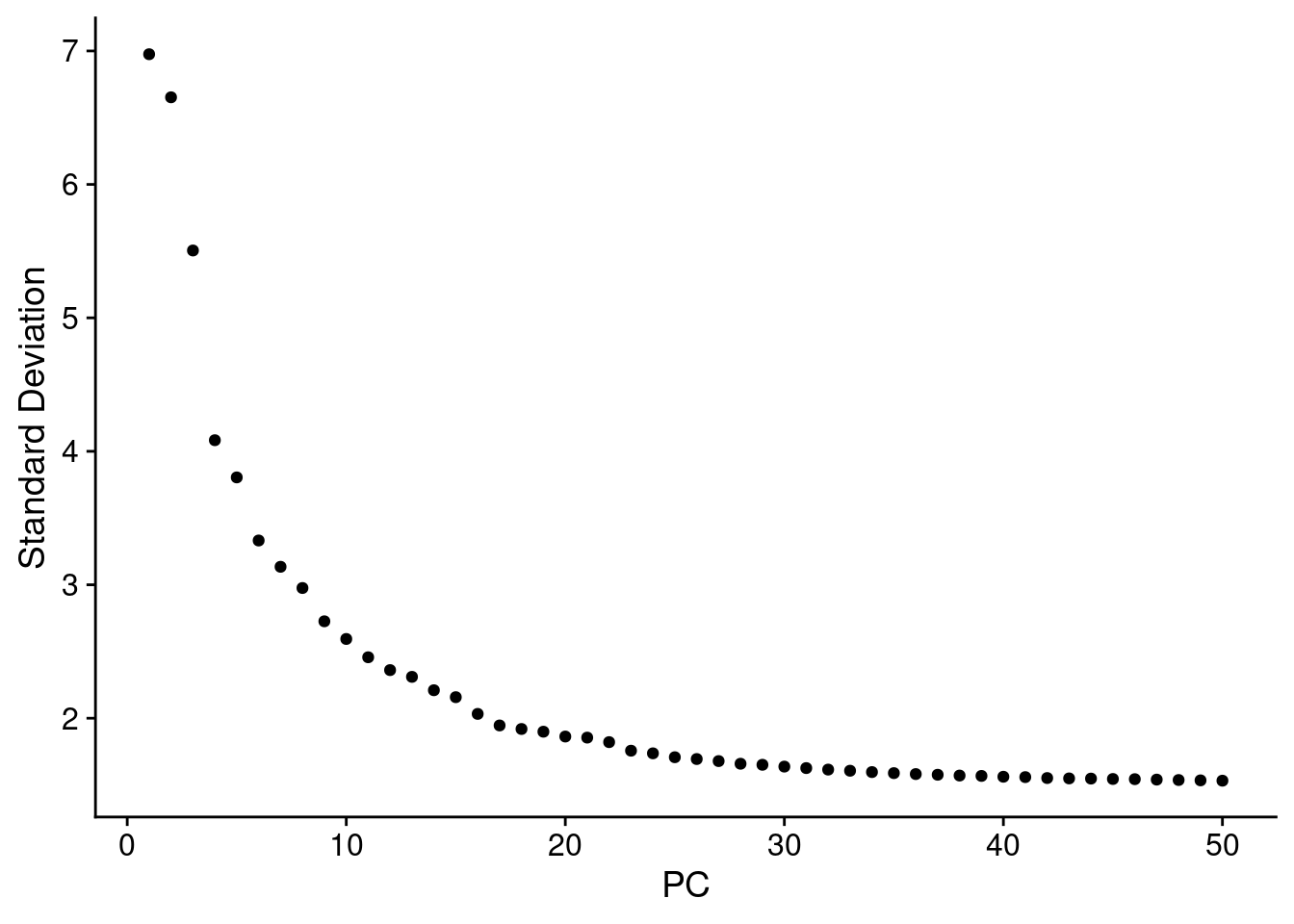
ElbowPlot (seurat.obj,ndims = 50 )
<- FindNeighbors (seurat.obj, dims = 1 : 25 )<- FindClusters (seurat.obj, resolution = 0.5 )
Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
Number of nodes: 4407
Number of edges: 148076
Running Louvain algorithm...
Maximum modularity in 10 random starts: 0.8695
Number of communities: 11
Elapsed time: 0 seconds
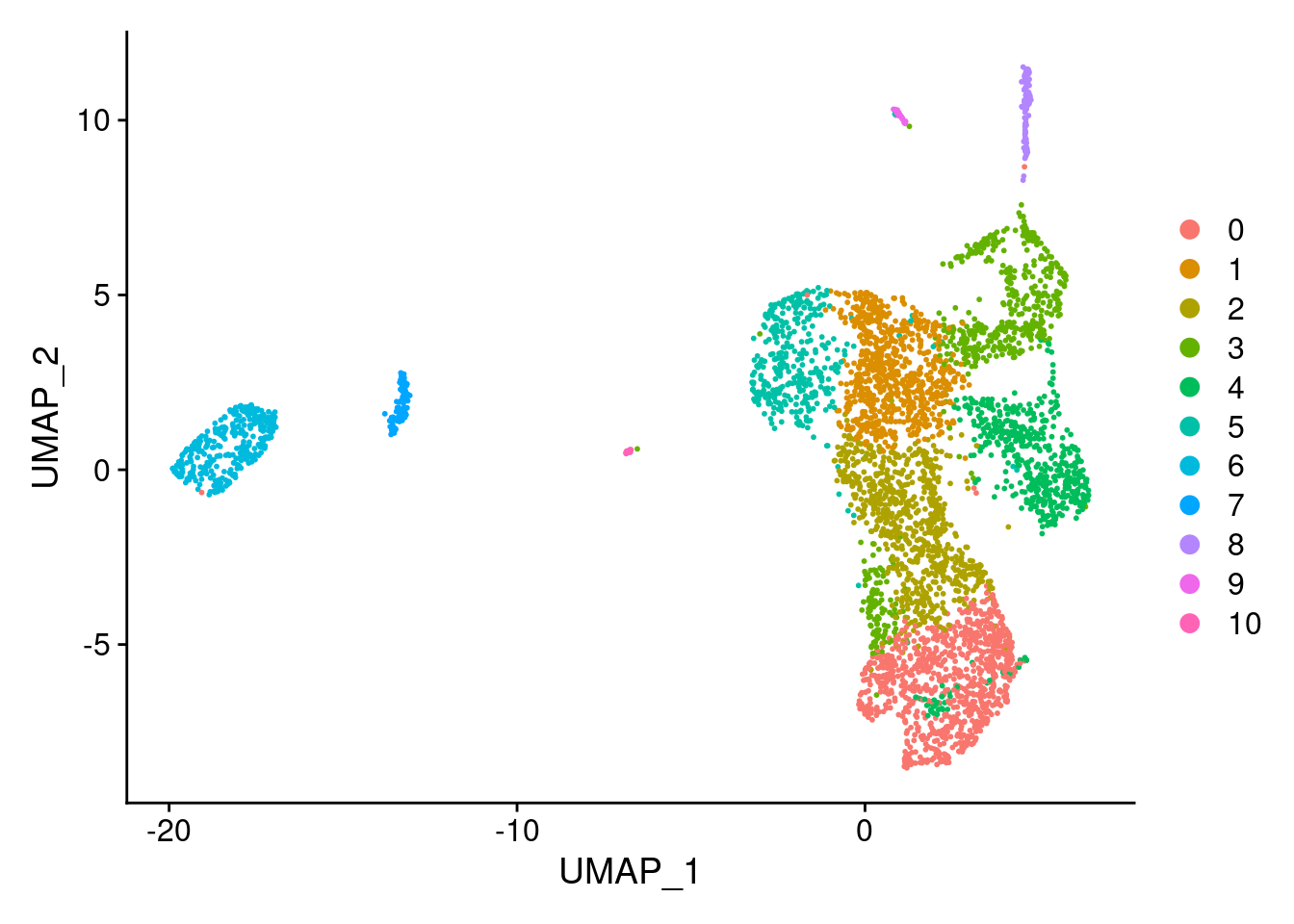
<- RunUMAP (seurat.obj, dims = 1 : 25 )
identical (rownames (seurat.obj@ meta.data),rownames (metaC))@ meta.data <- cbind (seurat.obj@ meta.data, metaC)DimPlot (seurat.obj,group.by = "Subclass" , label = TRUE )+ NoLegend ()
DimPlot (seurat.obj,group.by = "ClusterName" , label = T, label.size = 3 )+ NoLegend ()
<- SetIdent (seurat.obj,value = "Subclass" )<- FindAllMarkers (seurat.obj, logfc.threshold = 0.1 ,min.pct = 0.01 , only.pos = TRUE )head (seurat.obj.markers.Subclass)
p_val avg_log2FC pct.1 pct.2 p_val_adj
Neurod6 0.000000e+00 4.090494 0.987 0.127 0.000000e+00
Neurod2 3.164351e-294 3.376326 0.957 0.065 4.101949e-290
Tiam2 4.807385e-256 3.542872 0.900 0.068 6.231813e-252
Sox5 2.989319e-249 3.598904 0.912 0.127 3.875055e-245
Ppp2r2b 9.140509e-235 2.193826 0.970 0.361 1.184884e-230
Rbfox1 3.234905e-206 2.318090 0.974 0.412 4.193407e-202
cluster gene
Neurod6 Cortical or hippocampal glutamatergic Neurod6
Neurod2 Cortical or hippocampal glutamatergic Neurod2
Tiam2 Cortical or hippocampal glutamatergic Tiam2
Sox5 Cortical or hippocampal glutamatergic Sox5
Ppp2r2b Cortical or hippocampal glutamatergic Ppp2r2b
Rbfox1 Cortical or hippocampal glutamatergic Rbfox1
<- SetIdent (seurat.obj,value = "ClusterName" )<- FindAllMarkers (seurat.obj,densify = TRUE , logfc.threshold = 0.1 ,min.pct = 0.01 ,only.pos = TRUE )head (seurat.obj.markers.ClusterName)
p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
Wnt10a 4.836119e-259 0.9646853 0.316 0.000 6.269061e-255 Neur492 Wnt10a
Adamts19 2.737699e-201 0.8726592 0.316 0.001 3.548879e-197 Neur492 Adamts19
Tac2 1.894266e-172 2.0721515 0.316 0.002 2.455537e-168 Neur492 Tac2
Rmst 1.166934e-155 2.2966143 0.763 0.024 1.512697e-151 Neur492 Rmst
Eomes 1.978338e-139 1.6902540 0.579 0.015 2.564520e-135 Neur492 Eomes
Ebf1 4.915113e-109 1.9468755 0.526 0.016 6.371461e-105 Neur492 Ebf1
Comparision COTAN-Seurat
<- col_concat (crossing (colnames (dea.Subclass$ coex),c ("Seurat" ,"Cotan" )),sep = " " )<- vector ("list" , length (markers.list.names))names (markers.list) <- markers.list.names# I take positive coex significant genes for (cl.name in colnames (dea.Subclass$ coex)) {<- rownames (dea.Subclass$ coex[dea.Subclass$ ` p-value ` [,cl.name] < 0.01 & dea.Subclass$ coex[,cl.name] > 0 ,])paste0 (cl.name," Cotan" )]] <- genes#For seurat for (cl.name in colnames (dea.Subclass$ coex)) {<- seurat.obj.markers.Subclass[seurat.obj.markers.Subclass$ cluster == cl.name & seurat.obj.markers.Subclass$ p_val < 0.01 ,]$ genepaste0 (cl.name," Seurat" )]] <- genes
Cajal-Retzius subclass
<- ggVennDiagram (markers.list[1 : 2 ])
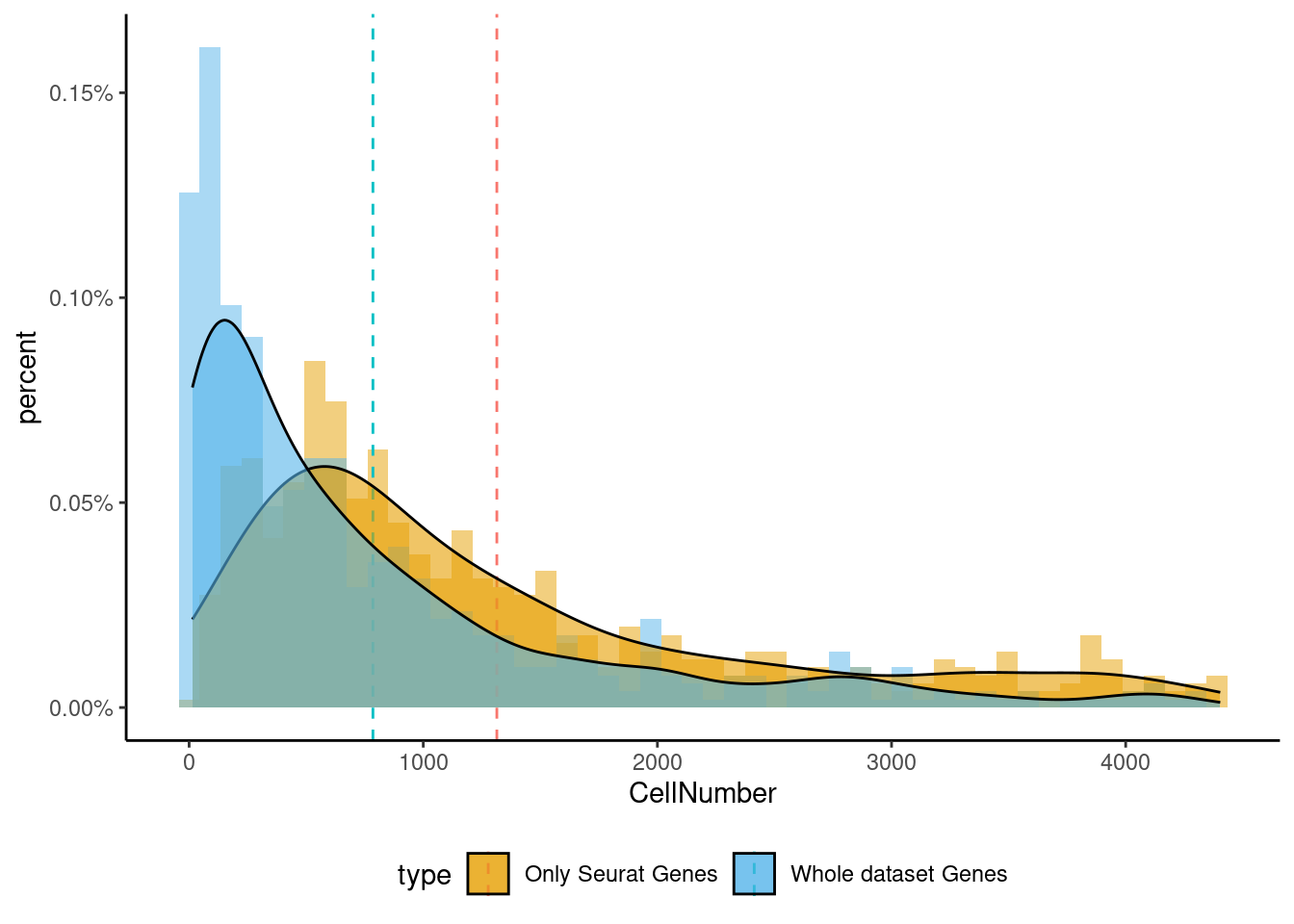
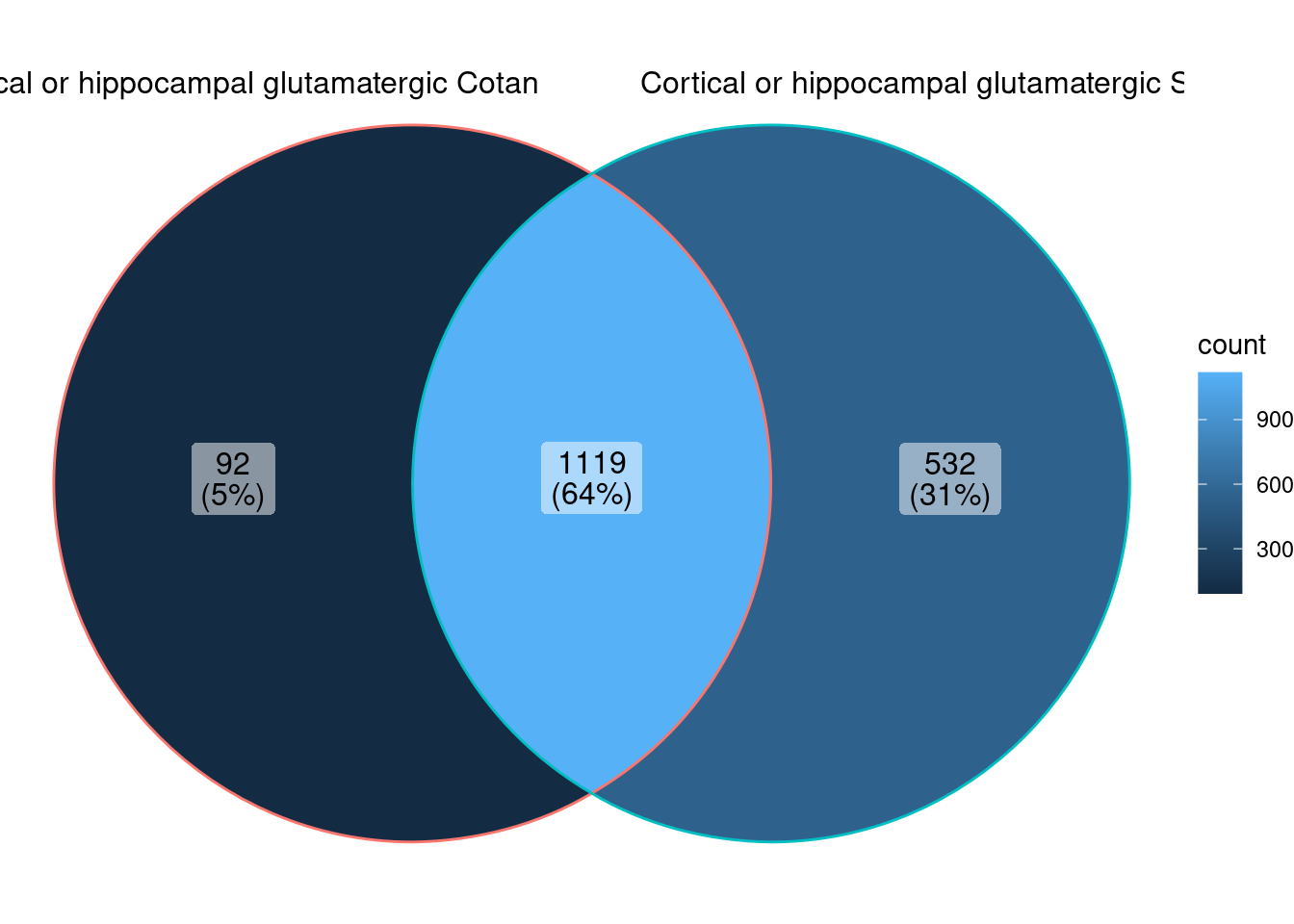
We can observe that there is a good overlap among the detected markers.
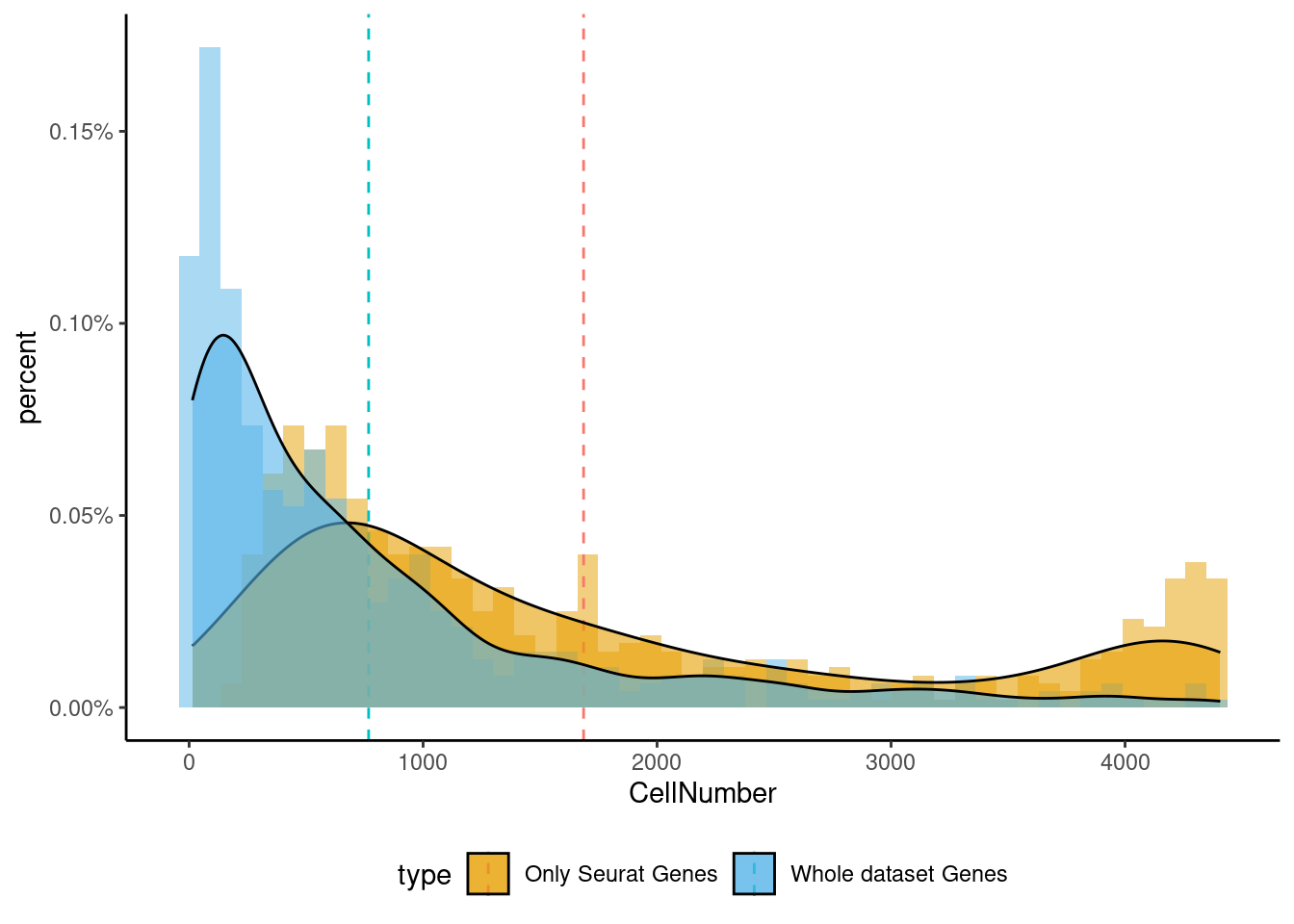
<- markers.list$ ` Cajal-Retzius Seurat ` [! markers.list$ ` Cajal-Retzius Seurat ` %in% markers.list$ ` Cajal-Retzius Cotan ` ]<- getNumOfExpressingCells (fb150Obj)[genes.to.test]<- as.data.frame (df)colnames (df) <- "CellNumber" rownames (df) <- NULL $ type <- "Only Seurat Genes" <- as.data.frame (getNumOfExpressingCells (fb150Obj)[sample (getGenes (fb150Obj), size = length (rownames (df)))])rownames (df.bk) <- NULL colnames (df.bk) <- "CellNumber" $ type <- "Whole dataset Genes" <- rbind (df,df.bk)<- ddply (df, "type" , summarise, grp.mean= mean (CellNumber))ggplot (df,aes (x= CellNumber,fill= type))+ scale_fill_manual (values= c ("#E69F00" , "#56B4E9" ))+ geom_histogram (aes (y= ..density..), position= "identity" , alpha= 0.5 ,bins = 50 )+ geom_vline (data= mu, aes (xintercept= grp.mean, color= type),linetype= "dashed" )+ geom_density (alpha= 0.6 )+ scale_y_continuous (labels = percent, name = "percent" ) + theme_classic ()+ theme (legend.position= "bottom" )
set.seed (111 )<- sample (genes.to.test,size = 12 )
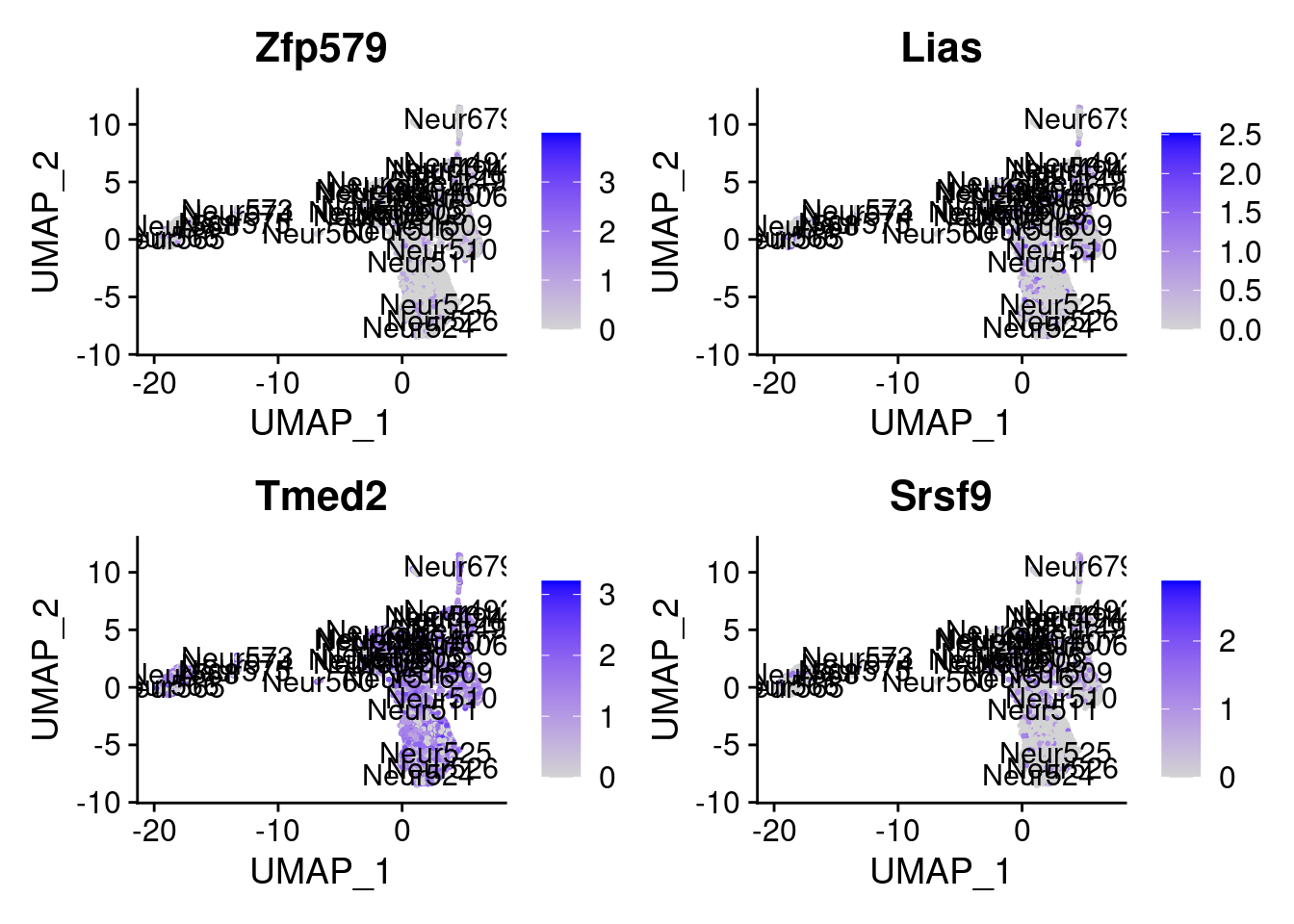
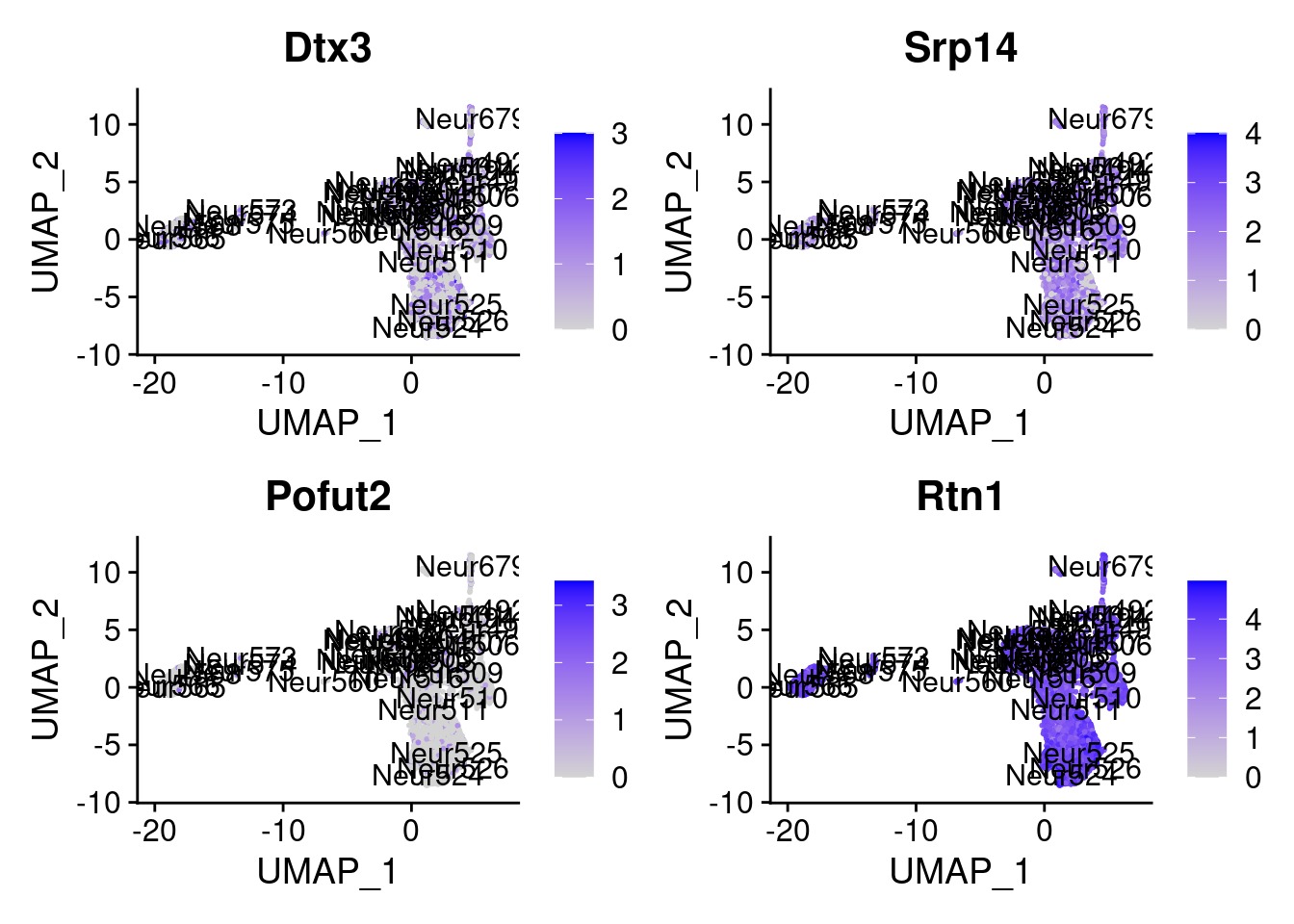
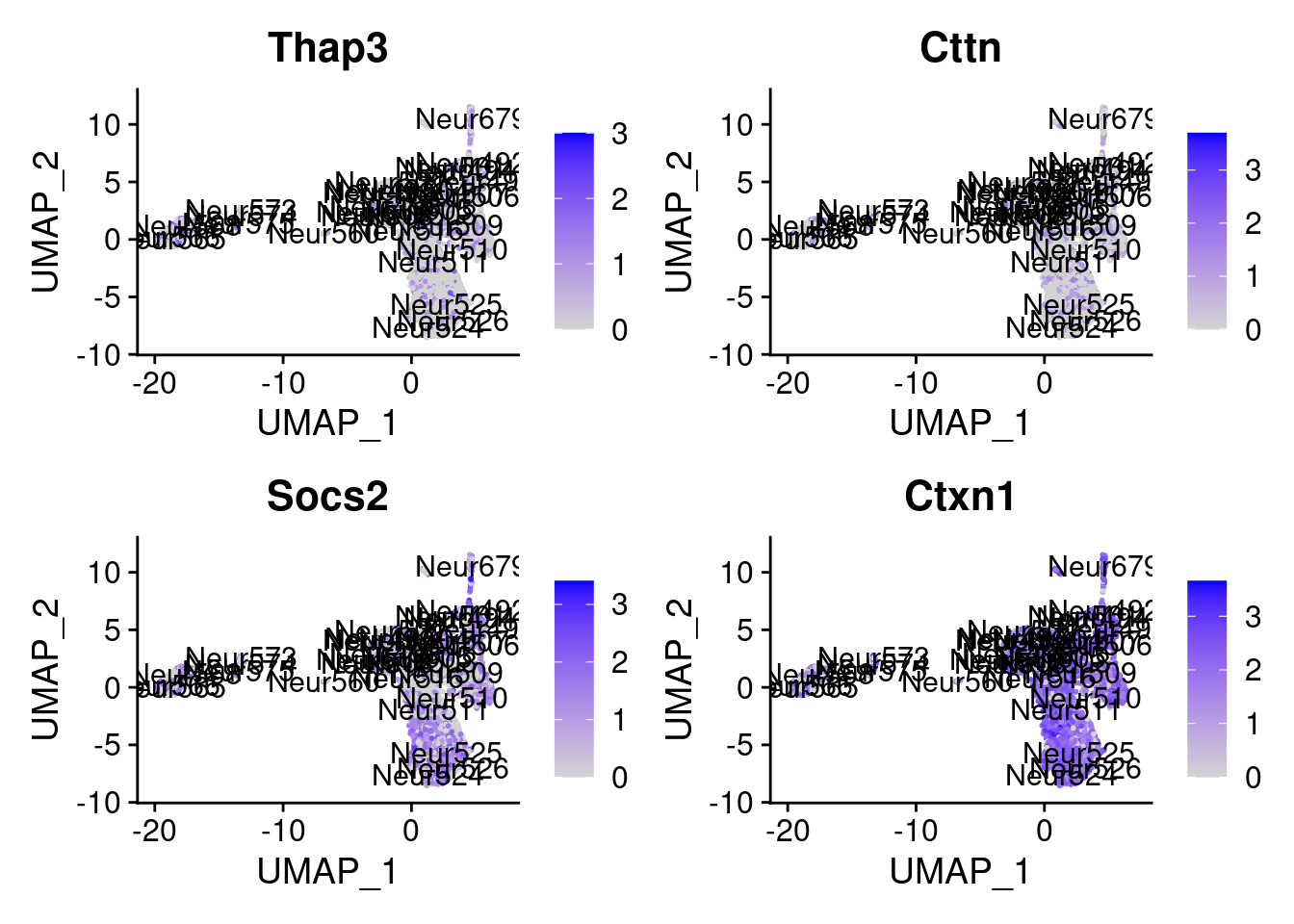
[1] "Zfp579" "Lias" "Tmed2" "Srsf9" "Dtx3" "Srp14" "Pofut2" "Rtn1"
[9] "Thap3" "Cttn" "Socs2" "Ctxn1"
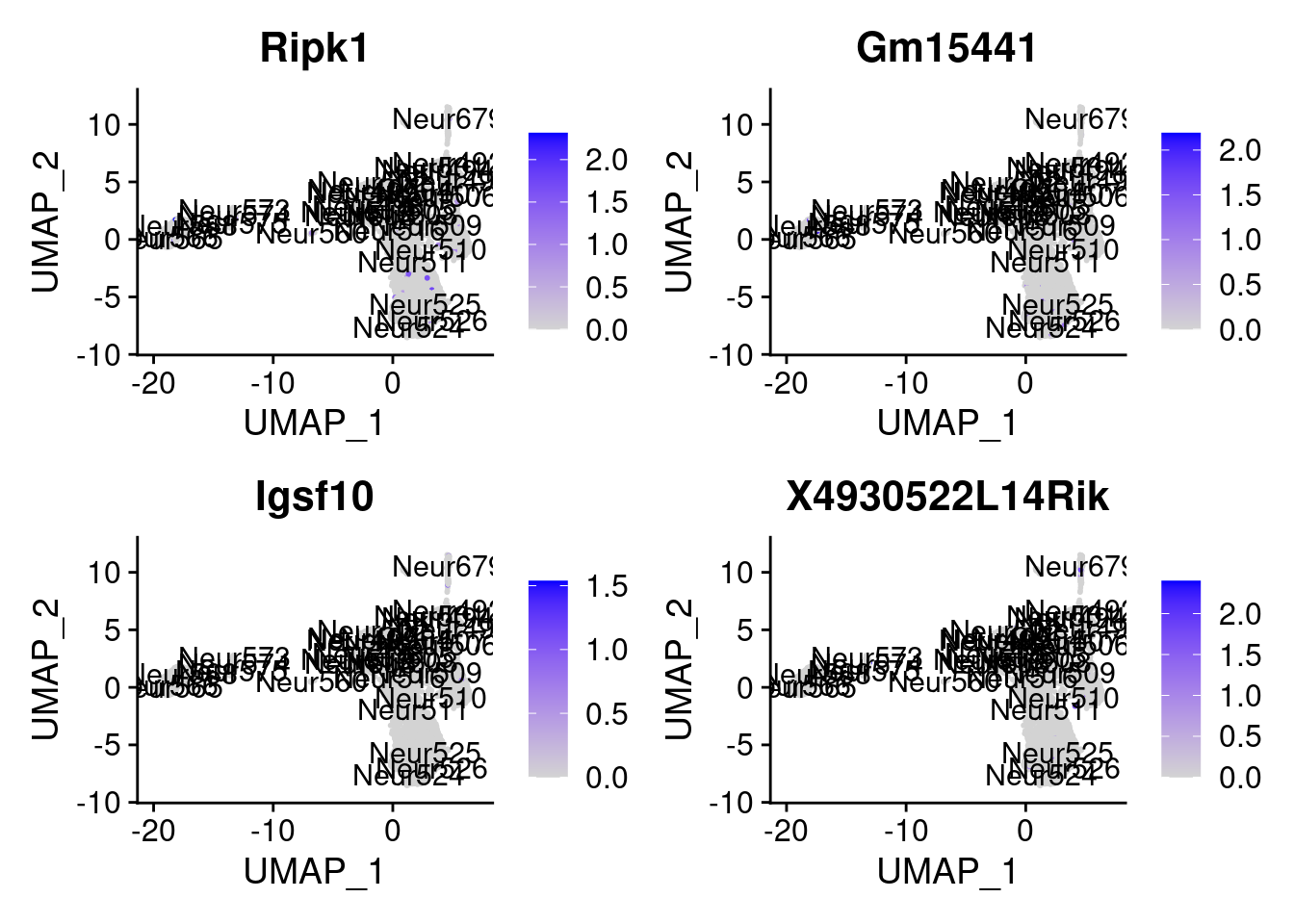
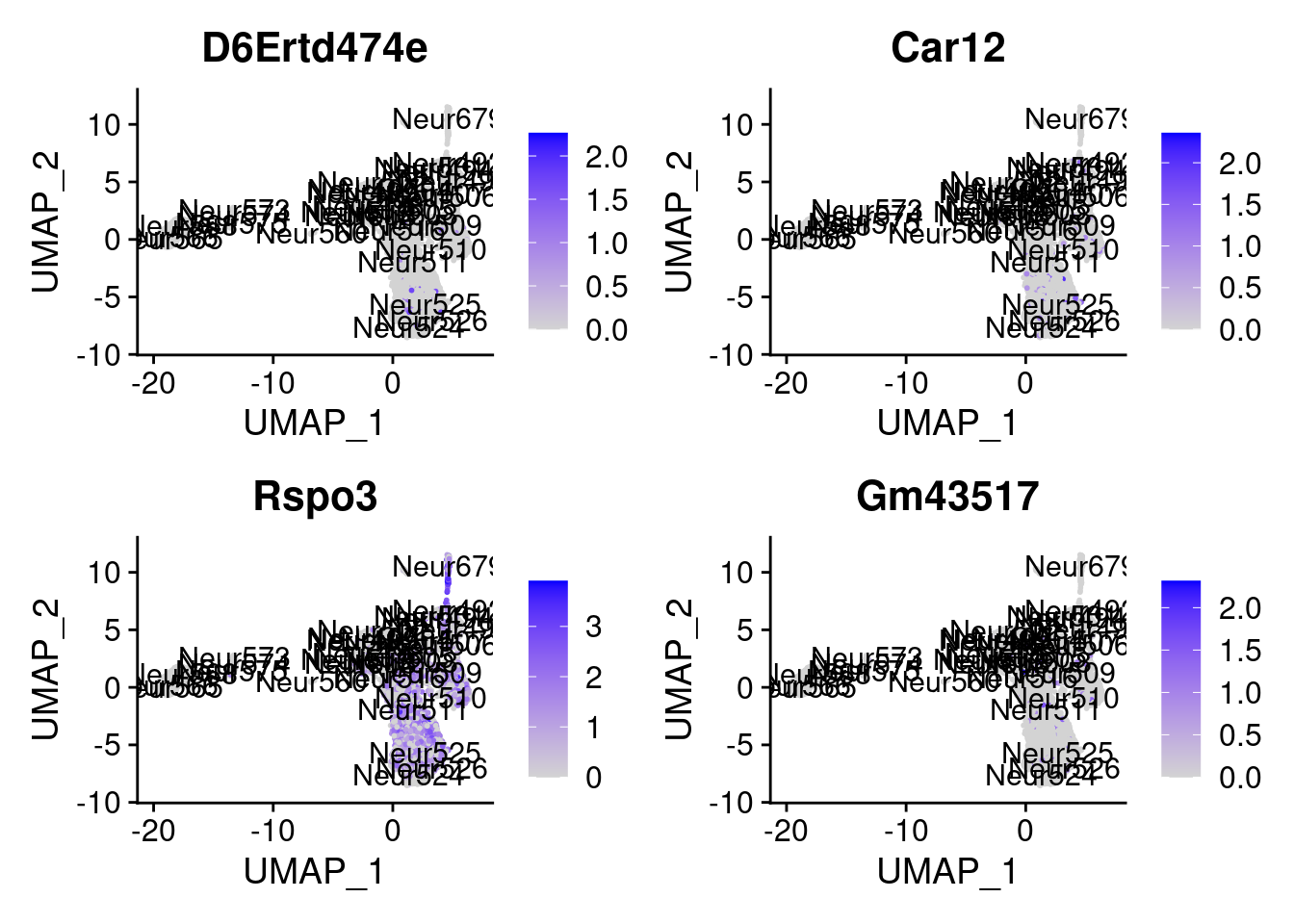
= 0 for (g in c (0 : 2 )) {= g* 4 plot (FeaturePlot (seurat.obj,features = genes[n+ c (1 : 4 )], label = T))
Generally if we look at the genes specifically detected by Seurat, they don’t seems so distinctive for CR cells.
sum (genes.to.test %in% markers.list$ ` Cortical or hippocampal glutamatergic Seurat ` )/ length (genes.to.test)
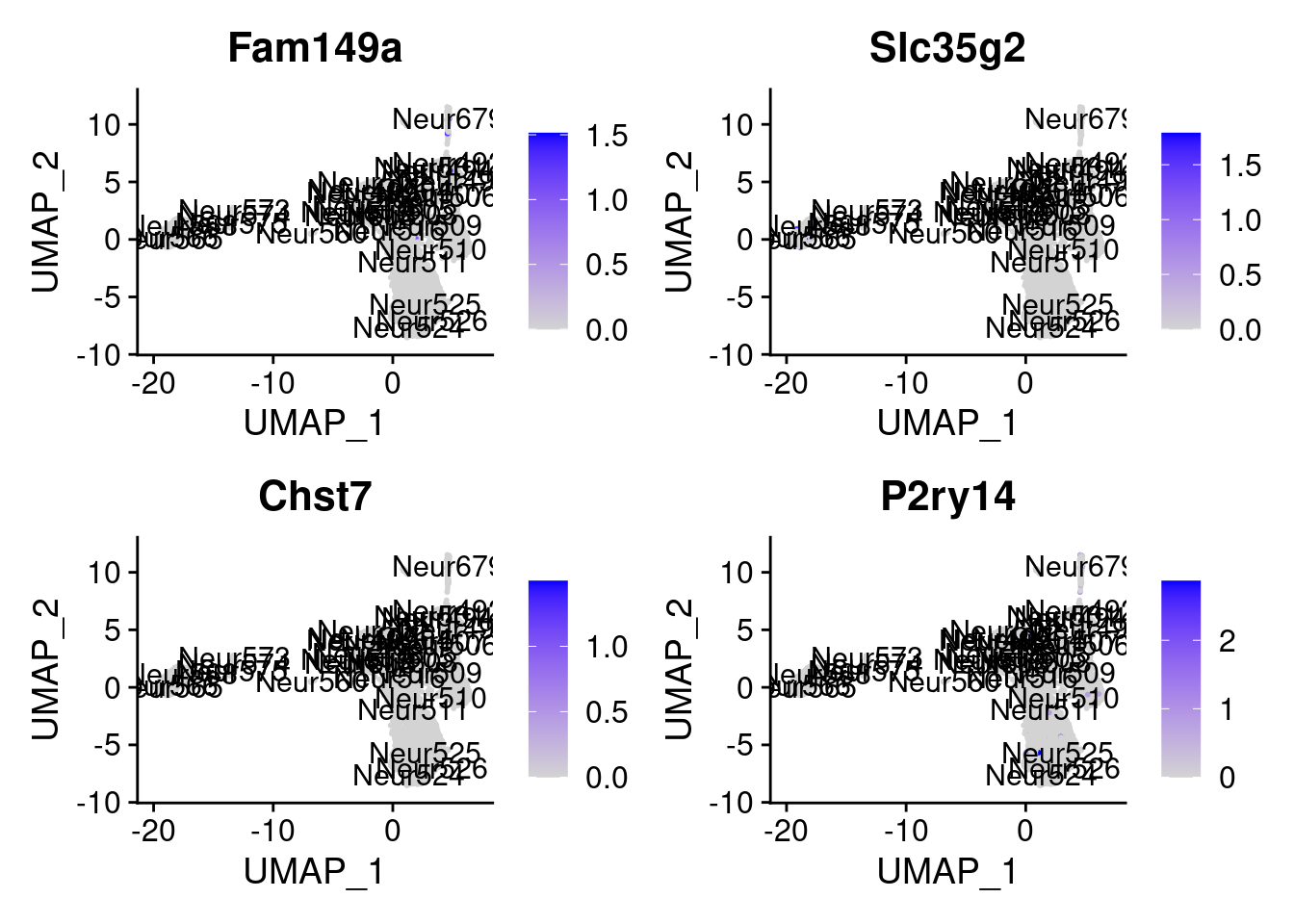
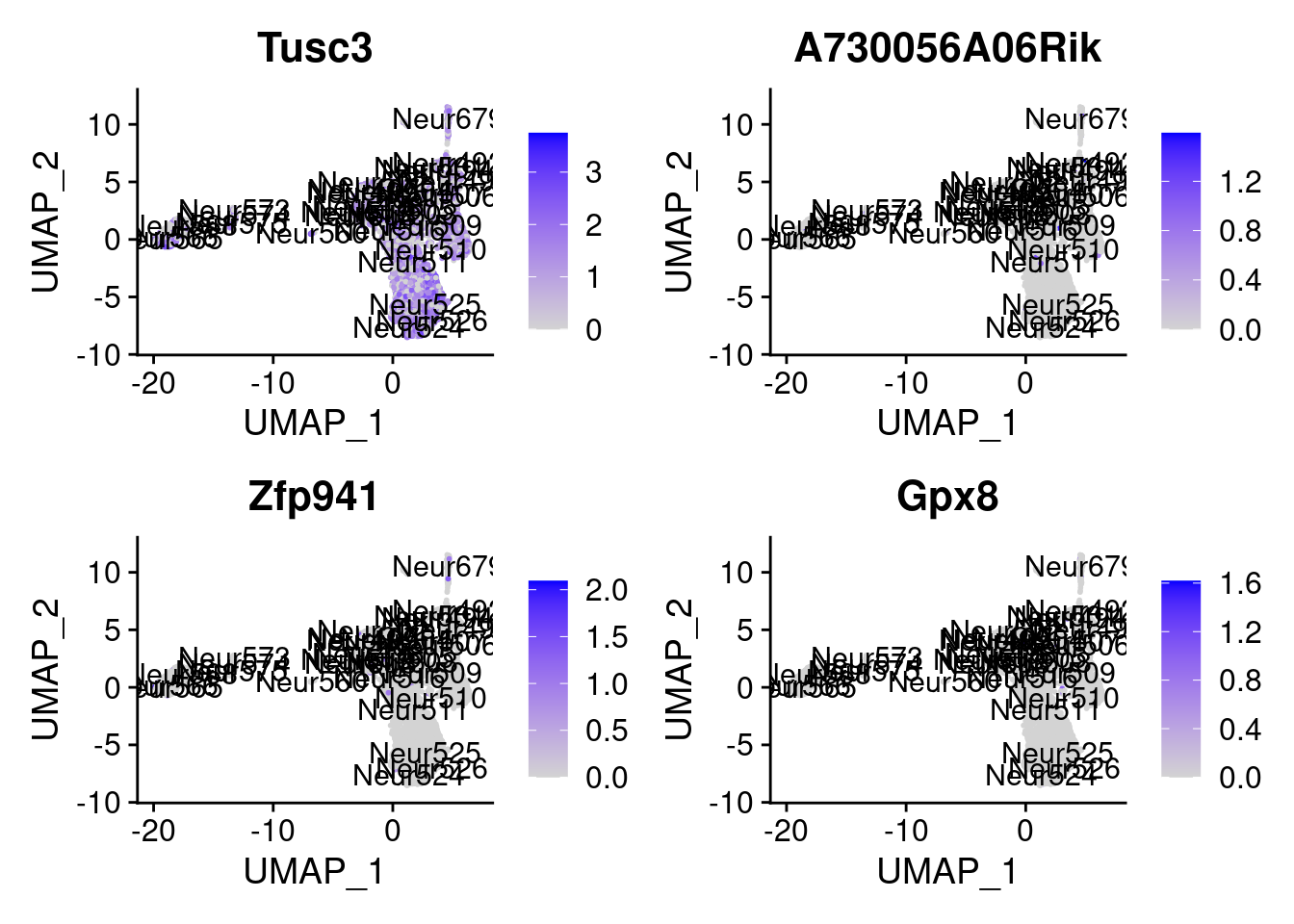
<- markers.list$ ` Cajal-Retzius Cotan ` [! markers.list$ ` Cajal-Retzius Cotan ` %in% markers.list$ ` Cajal-Retzius Seurat ` ]set.seed (11 )<- sample (genes.to.test.Cotan,size = 30 )
[1] "Fam149a" "Slc35g2" "Chst7" "P2ry14"
[5] "Tusc3" "A730056A06Rik" "Zfp941" "Gpx8"
[9] "Ripk1" "Gm15441" "Igsf10" "X4930522L14Rik"
[13] "Gm16845" "Gspt2" "Scn2b" "Akr1b10"
[17] "Vegfa" "X4933431E20Rik" "Rtkn" "Gm38250"
[21] "Fancf" "Rab43" "Grik1" "Eya1"
[25] "Pih1d2" "Cldn12" "Cacng2" "Pias3"
[29] "Gm26782" "Mme"
= 0 for (g in c (0 : 2 )) {= g* 4 plot (FeaturePlot (seurat.obj,features = genes[n+ c (1 : 4 )], label = T))
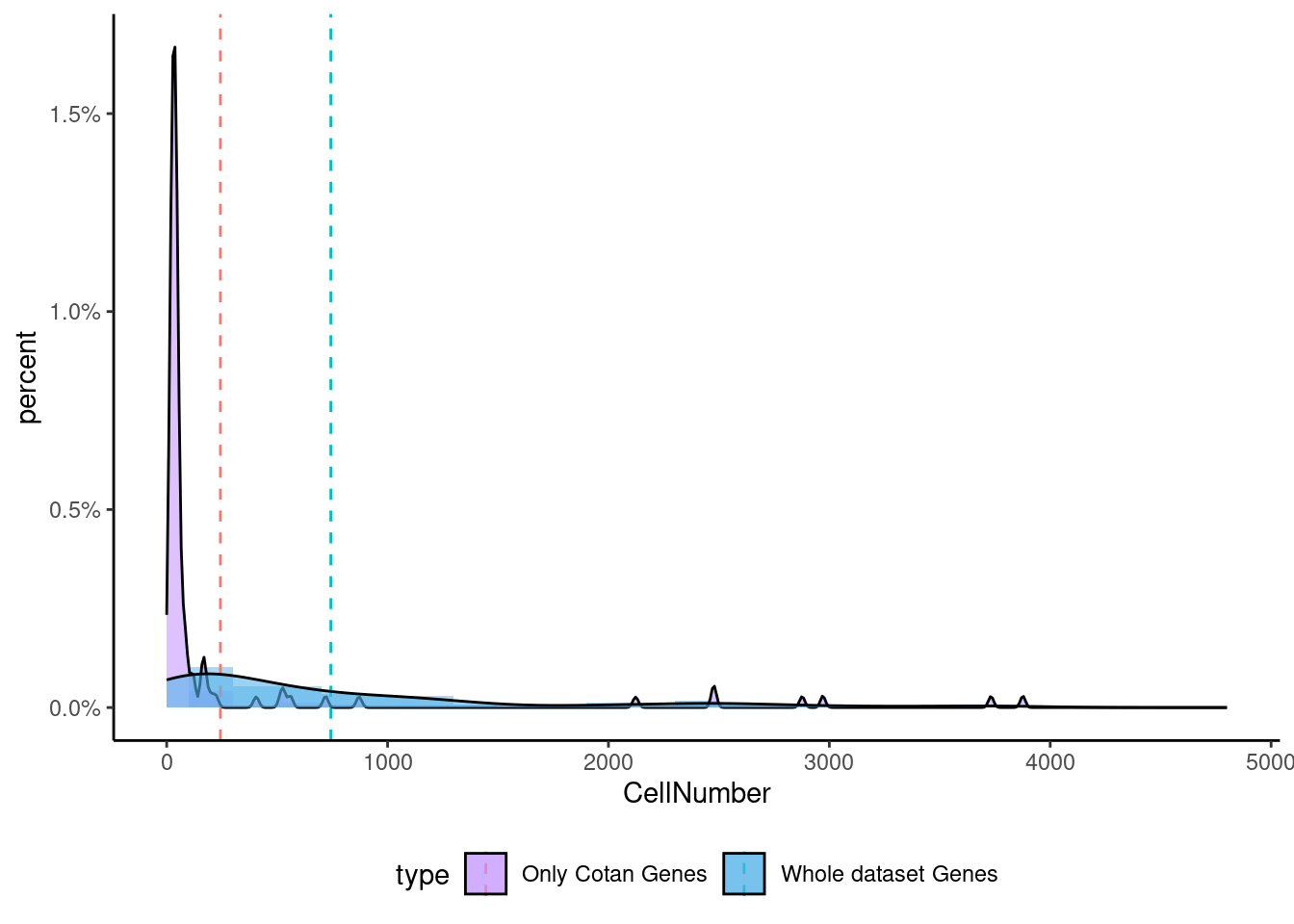
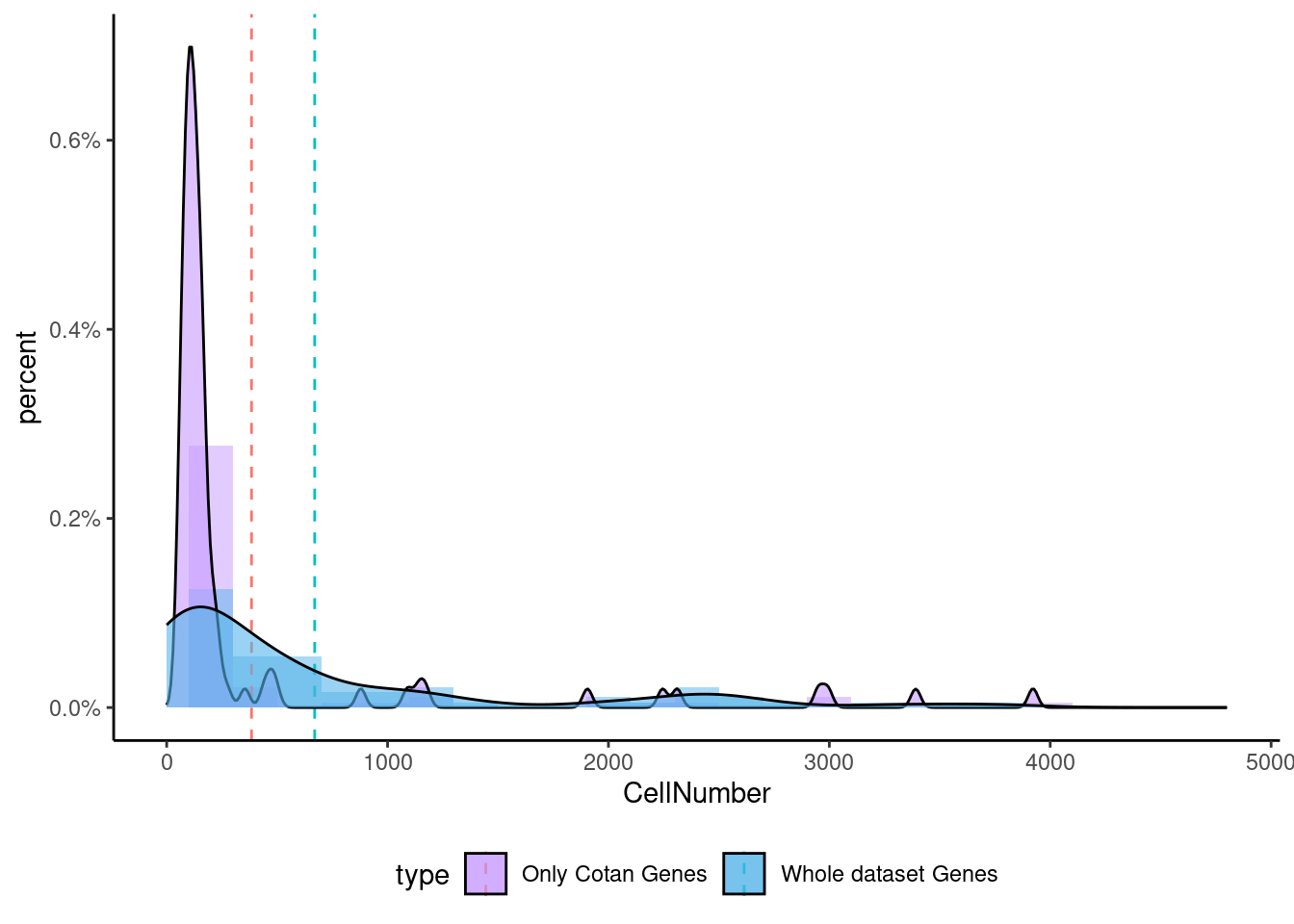
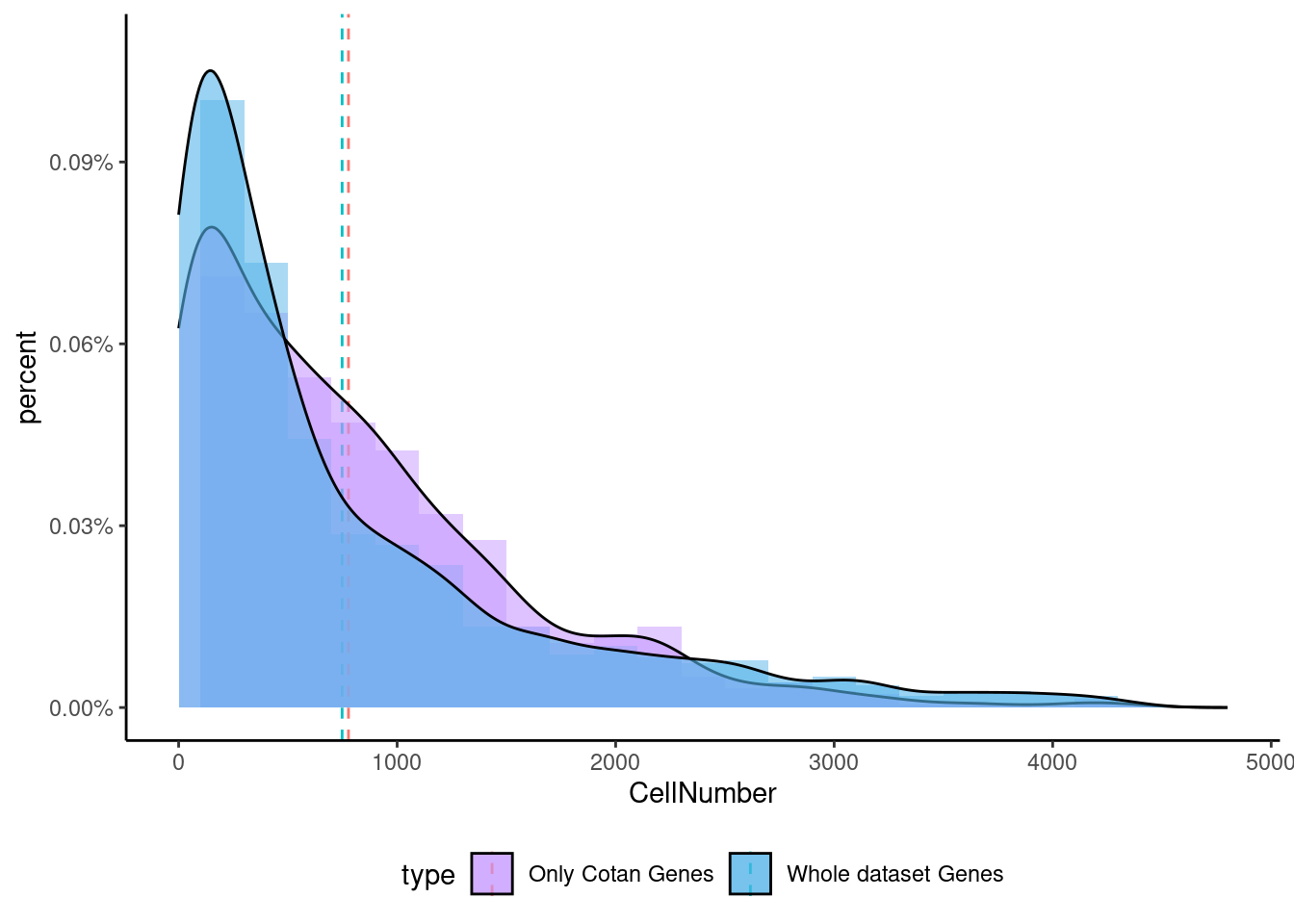
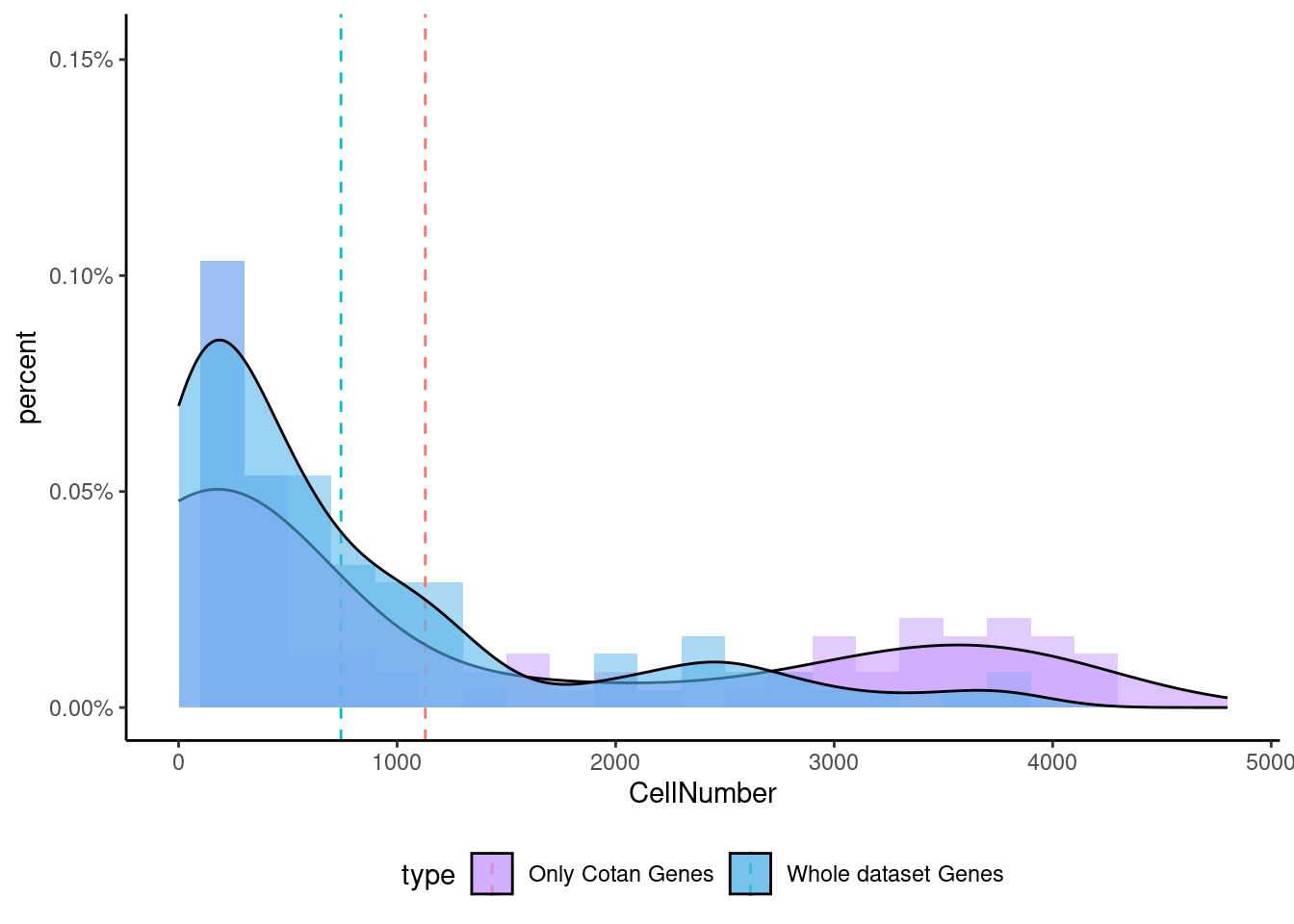
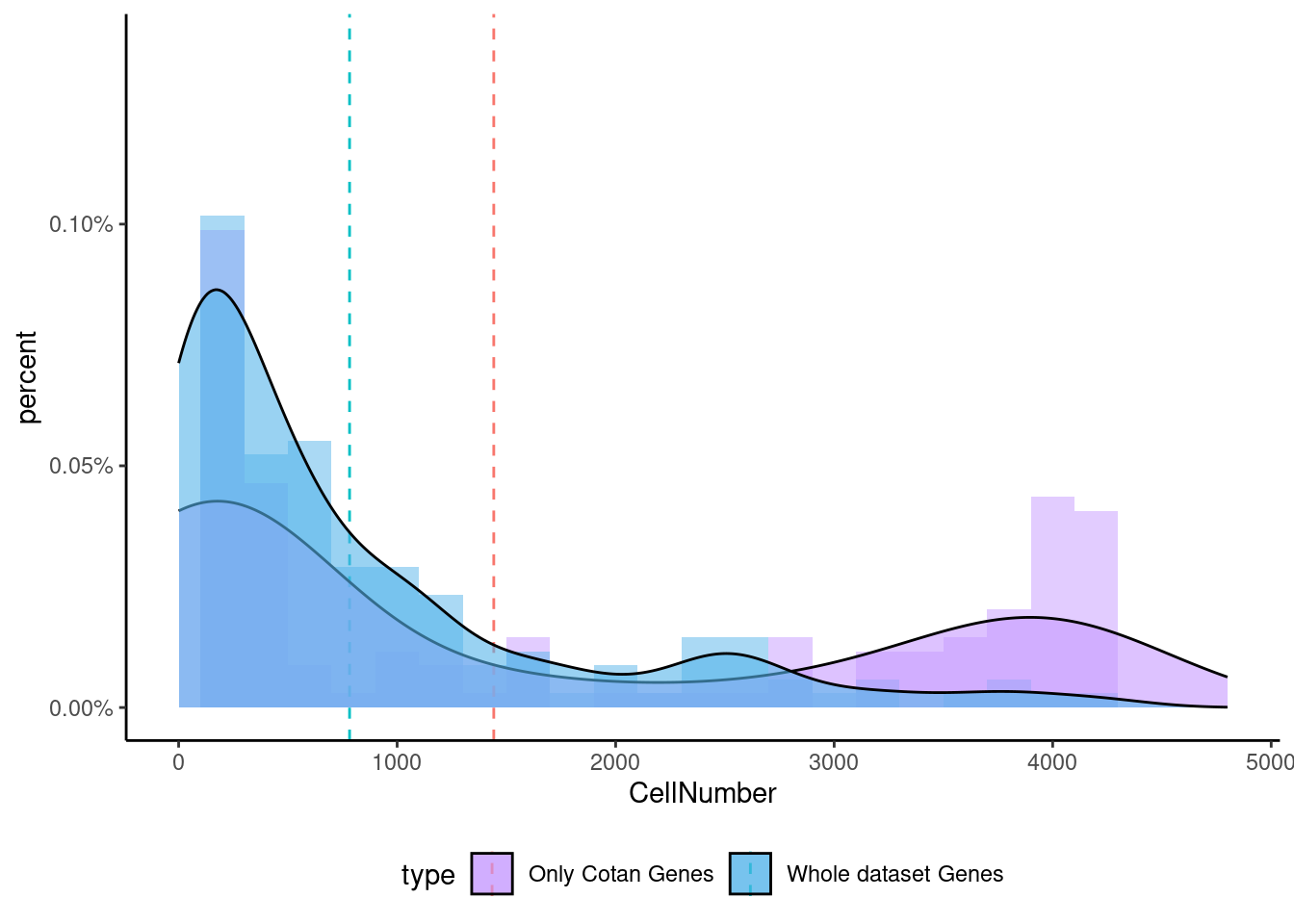
<- getNumOfExpressingCells (fb150Obj)[genes.to.test.Cotan]<- as.data.frame (df)colnames (df) <- "CellNumber" rownames (df) <- NULL $ type <- "Only Cotan Genes" <- as.data.frame (getNumOfExpressingCells (fb150Obj)[sample (getGenes (fb150Obj), size = length (rownames (df)))])rownames (df.bk) <- NULL colnames (df.bk) <- "CellNumber" $ type <- "Whole dataset Genes" <- rbind (df,df.bk)<- ddply (df, "type" , summarise, grp.mean= mean (CellNumber))ggplot (df,aes (x= CellNumber,fill= type))+ scale_fill_manual (values= c ("#C69AFF" , "#56B4E9" ))+ geom_histogram (aes (y= ..density..), position= "identity" , alpha= 0.5 ,bins = 25 )+ xlim (0 ,4800 )+ geom_vline (data= mu, aes (xintercept= grp.mean, color= type),linetype= "dashed" )+ geom_density (alpha= 0.6 )+ scale_y_continuous (labels = percent, name = "percent" ) + theme_classic ()+ theme (legend.position= "bottom" )
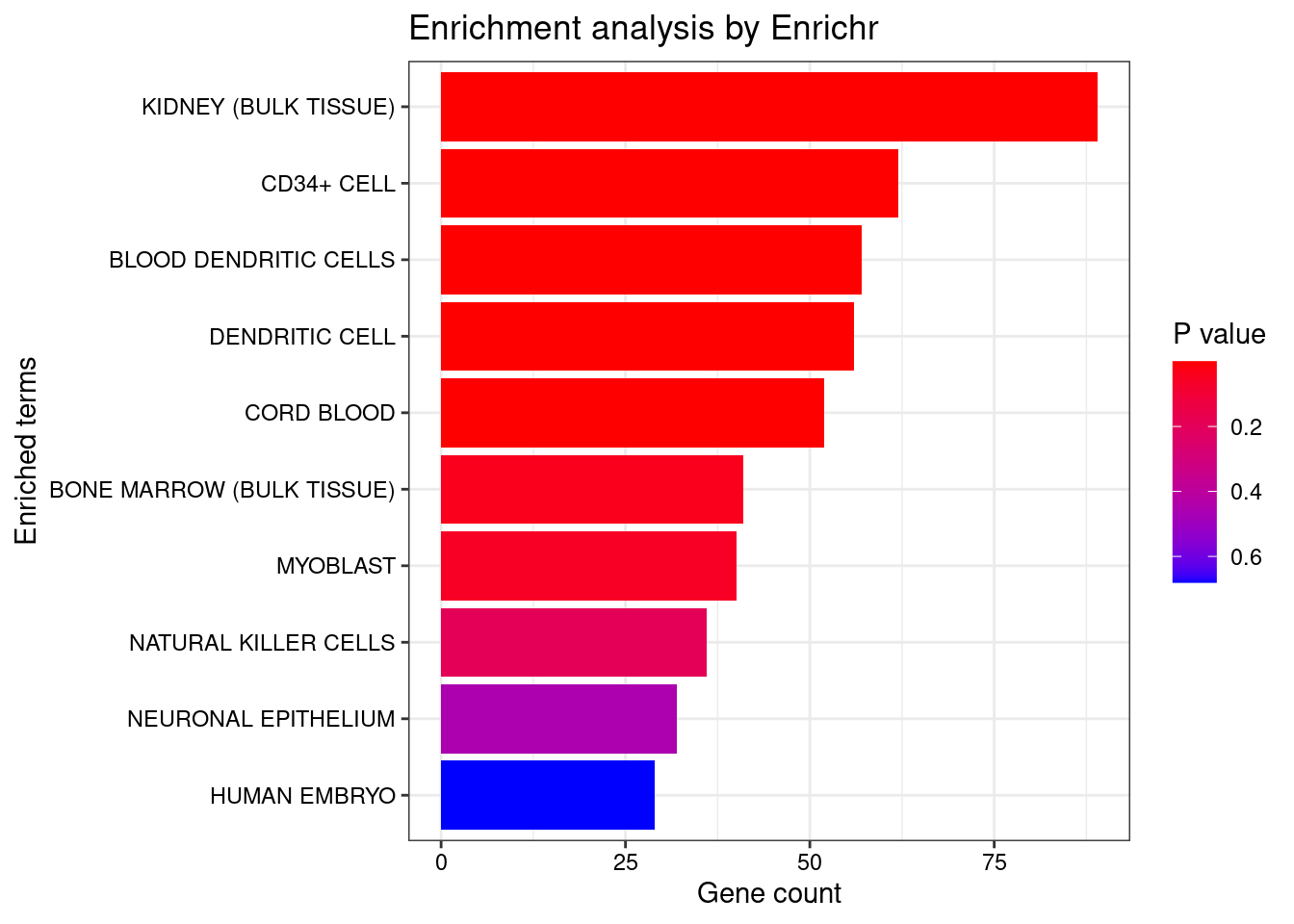
Now we can test the enrichment for the specific gene both in Seurat and in Cotan.
To make a comparison more equal we select the same number of genes (depending on the smallest group)
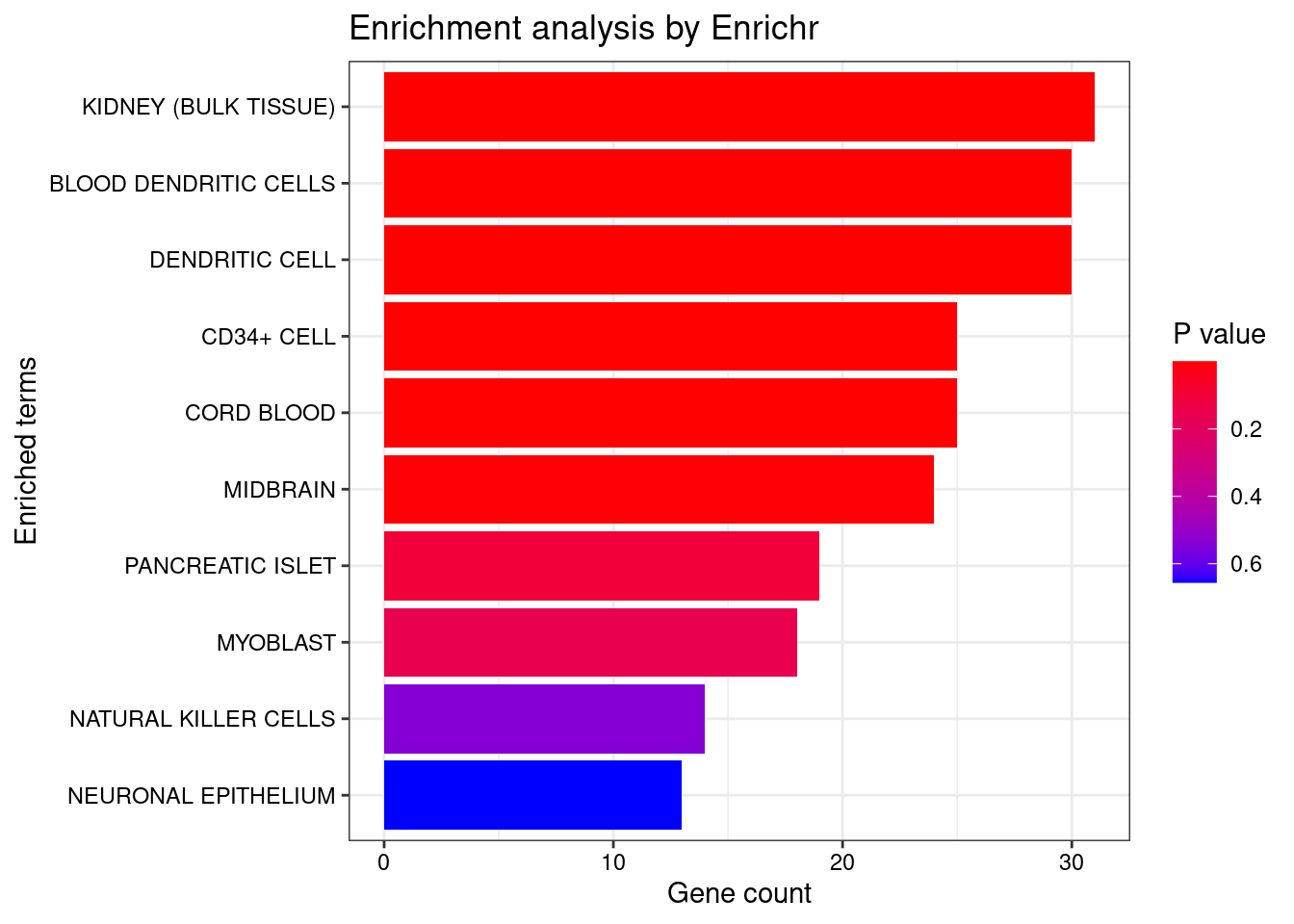
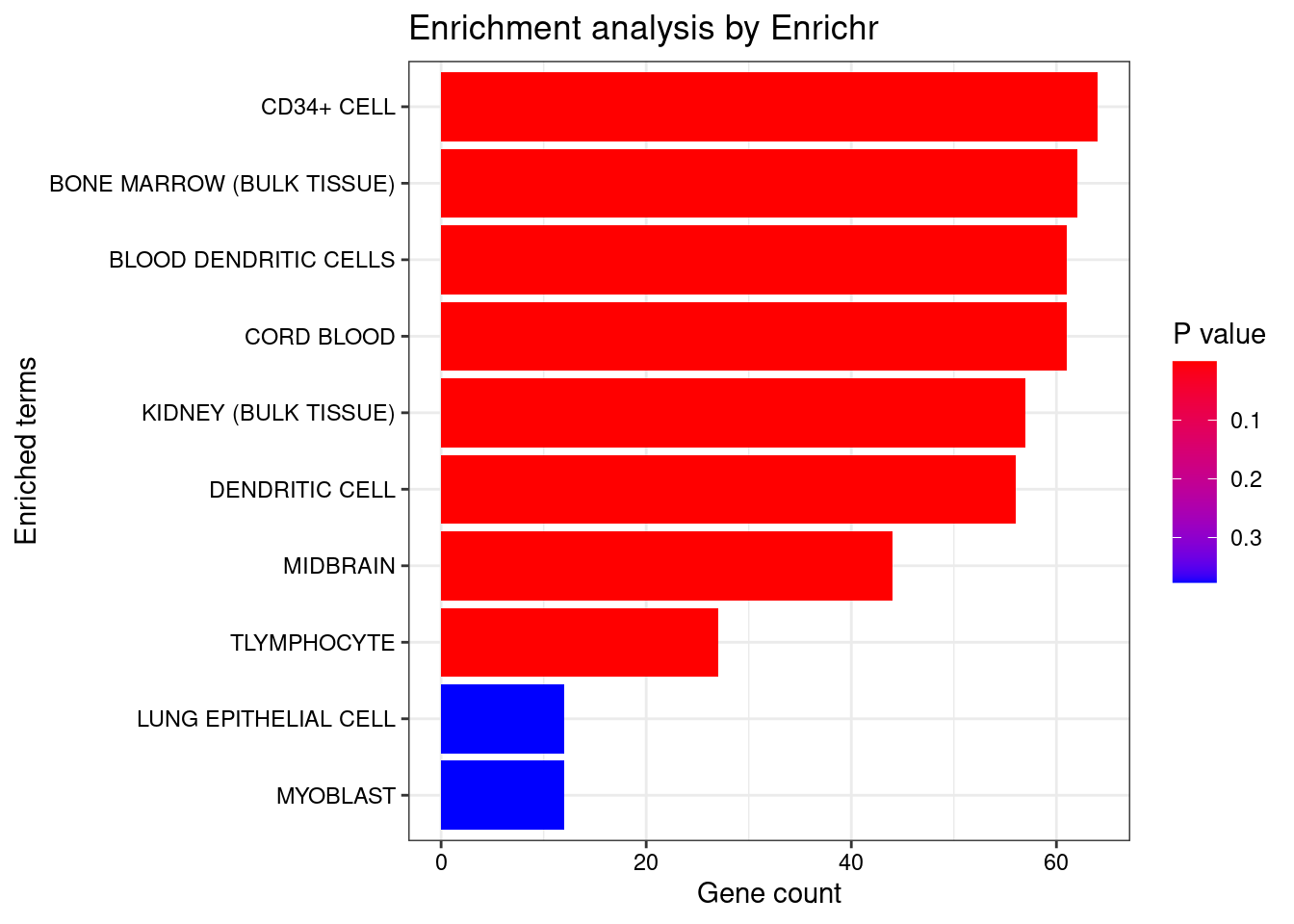
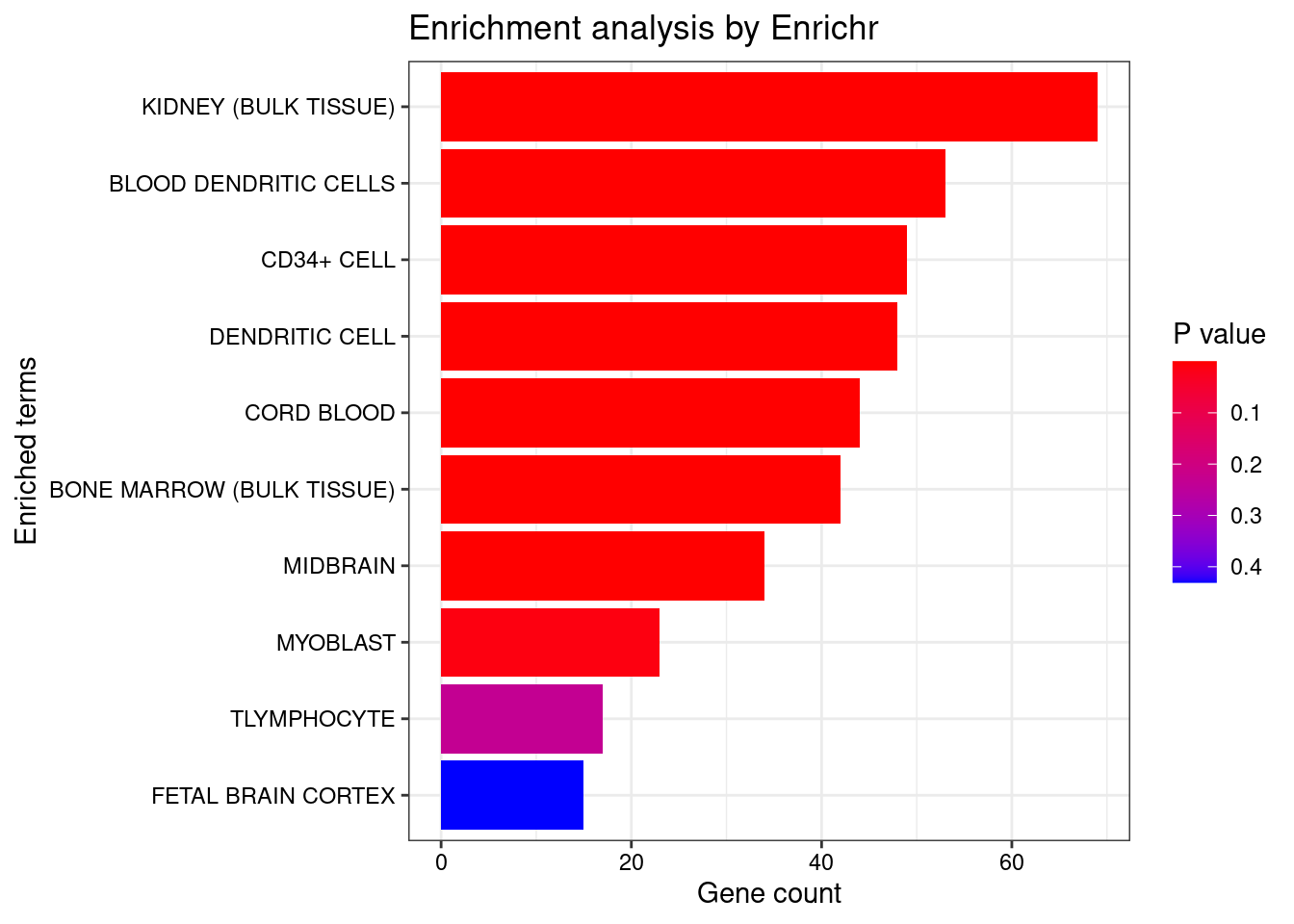
<- seurat.obj.markers.Subclass[seurat.obj.markers.Subclass$ cluster == "Cajal-Retzius" & seurat.obj.markers.Subclass$ gene %in% genes.to.test,]$ gene[1 : length (genes.to.test.Cotan)]<- enrichr (genes.to.testTop, dbs)
Uploading data to Enrichr... Done.
Querying ARCHS4_Tissues... Done.
Parsing results... Done.
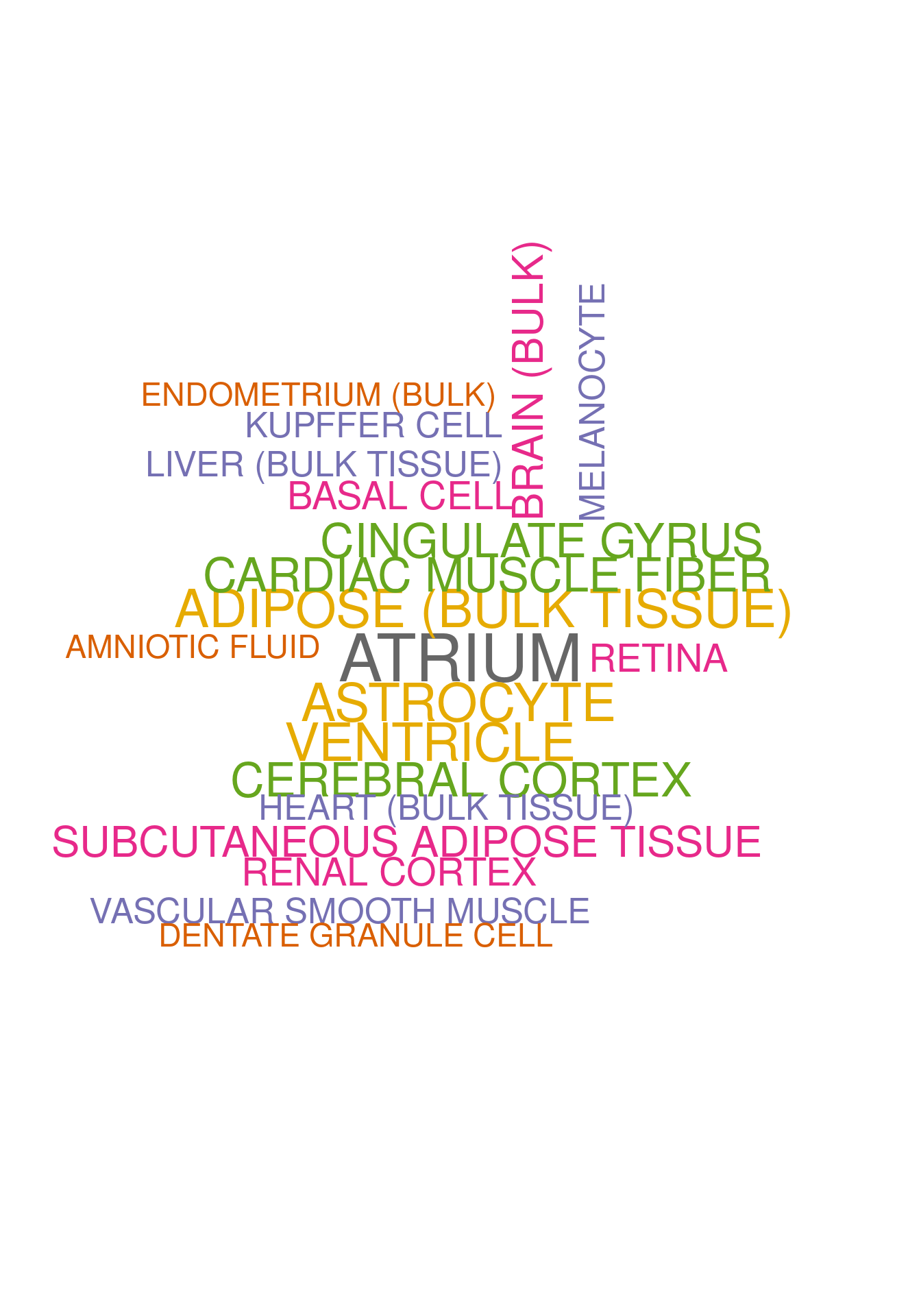
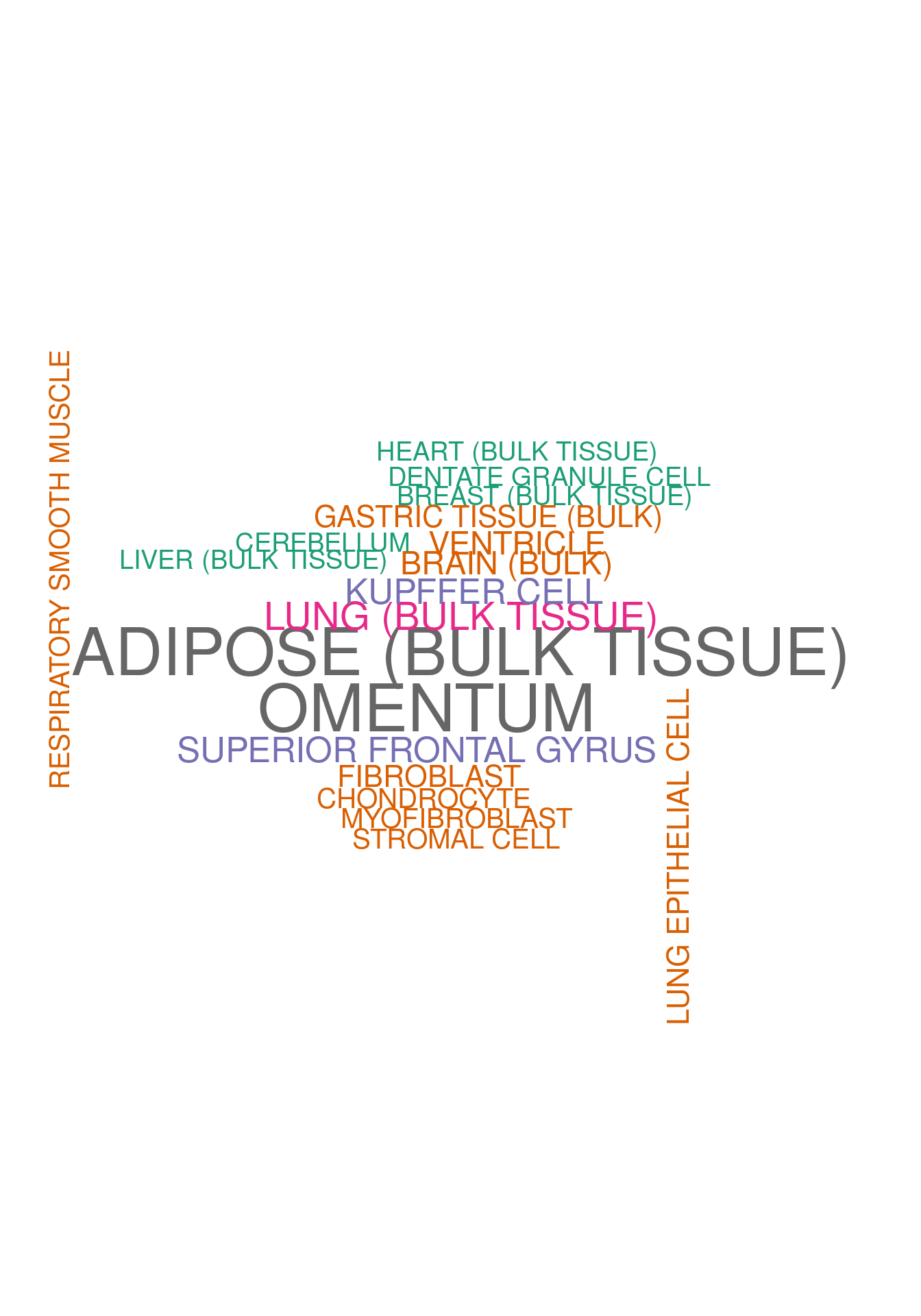
plotEnrich (enriched[[1 ]], showTerms = 10 , numChar = 40 , y = "Count" , orderBy = "P.value" )
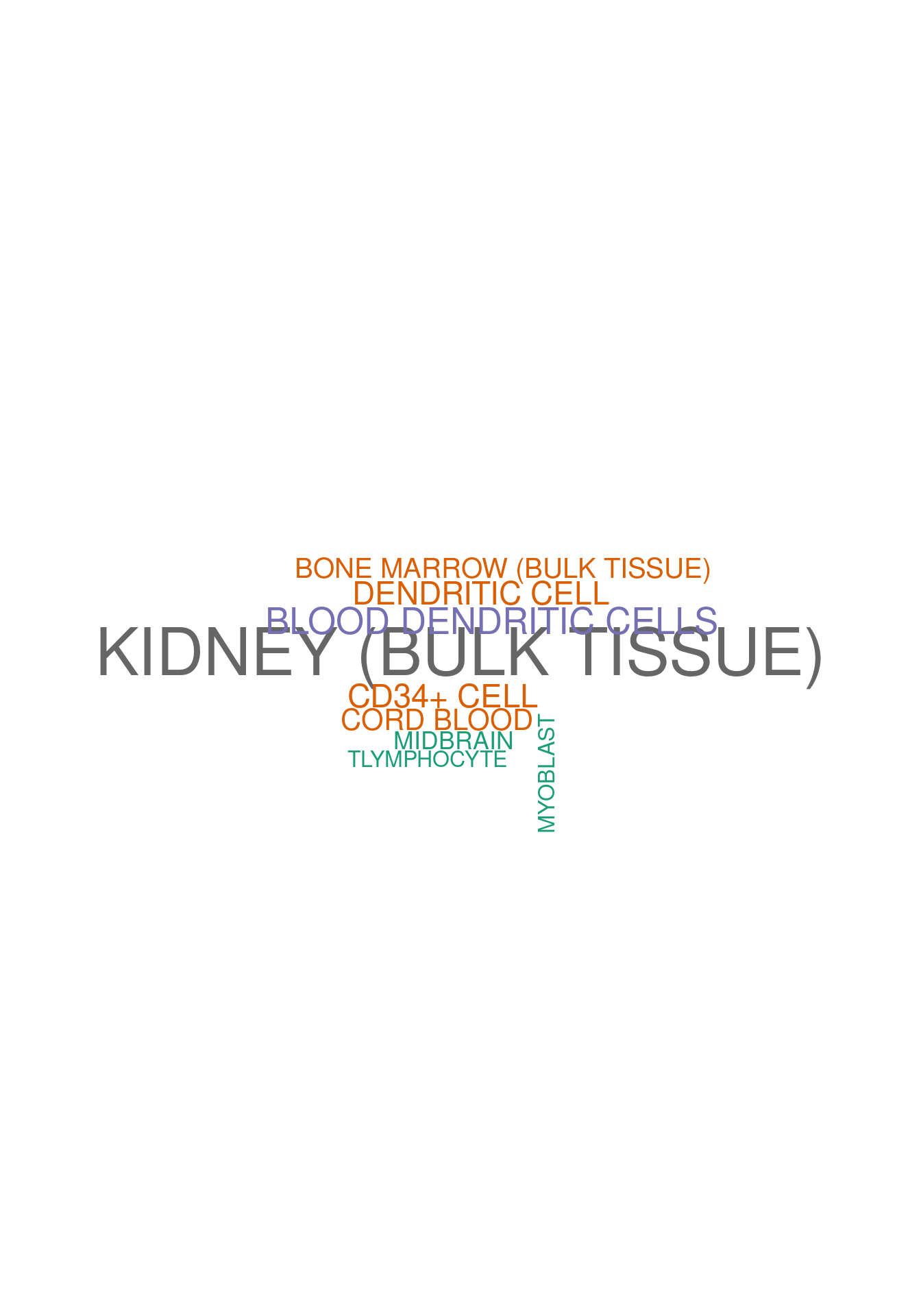
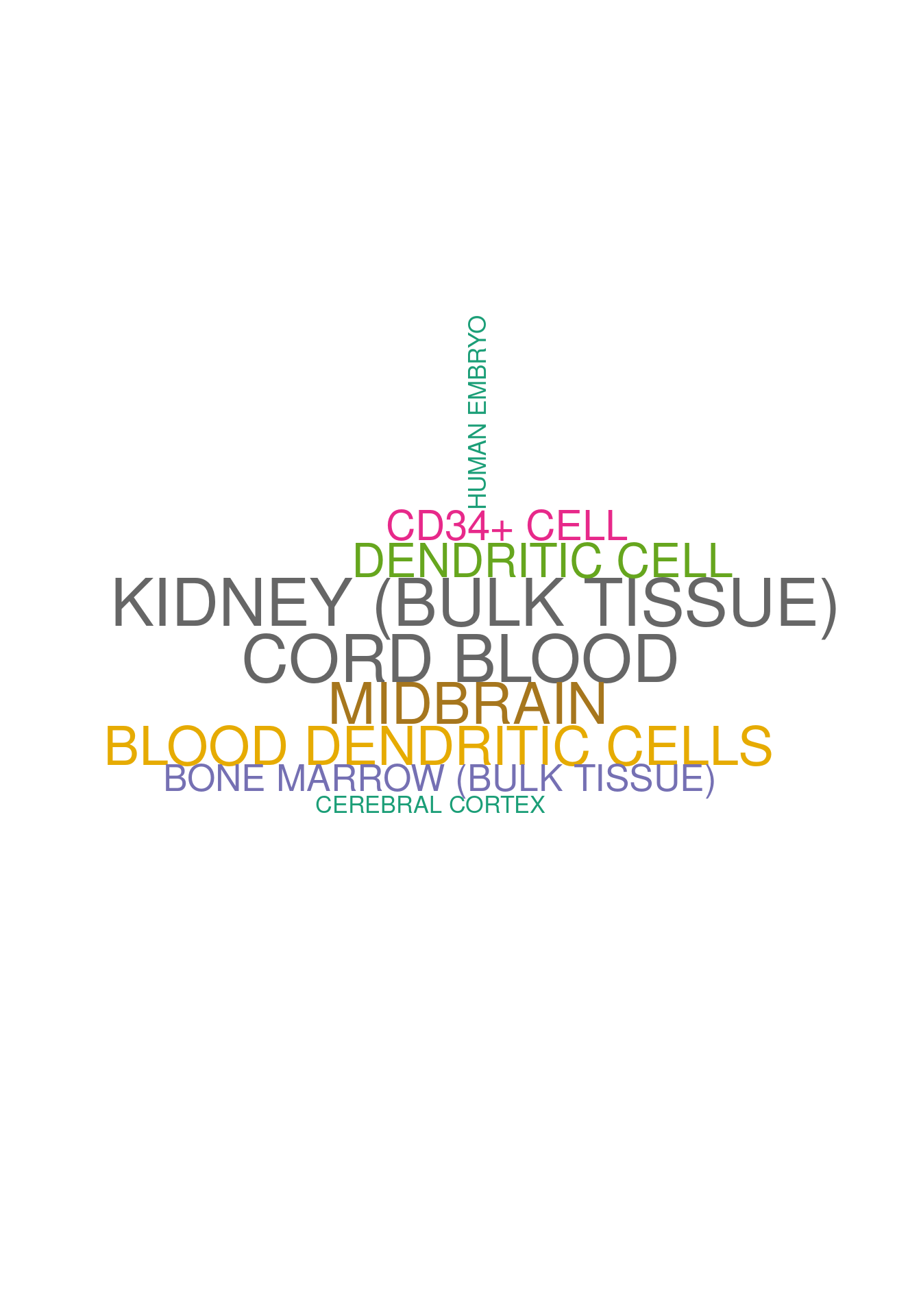
set.seed (123 )<- data.frame (Terms = str_split (enriched[[1 ]]$ Term[1 : 20 ],pattern = " CL" ,simplify = T )[,1 ],Scores = enriched[[1 ]]$ Combined.Score[1 : 20 ])wordcloud (wordcloud_data$ Terms, wordcloud_data$ Scores, scale = c (3 , 1 ), min.freq = 1 , random.order= FALSE , rot.per= 0.1 ,colors= brewer.pal (8 , "Dark2" ))
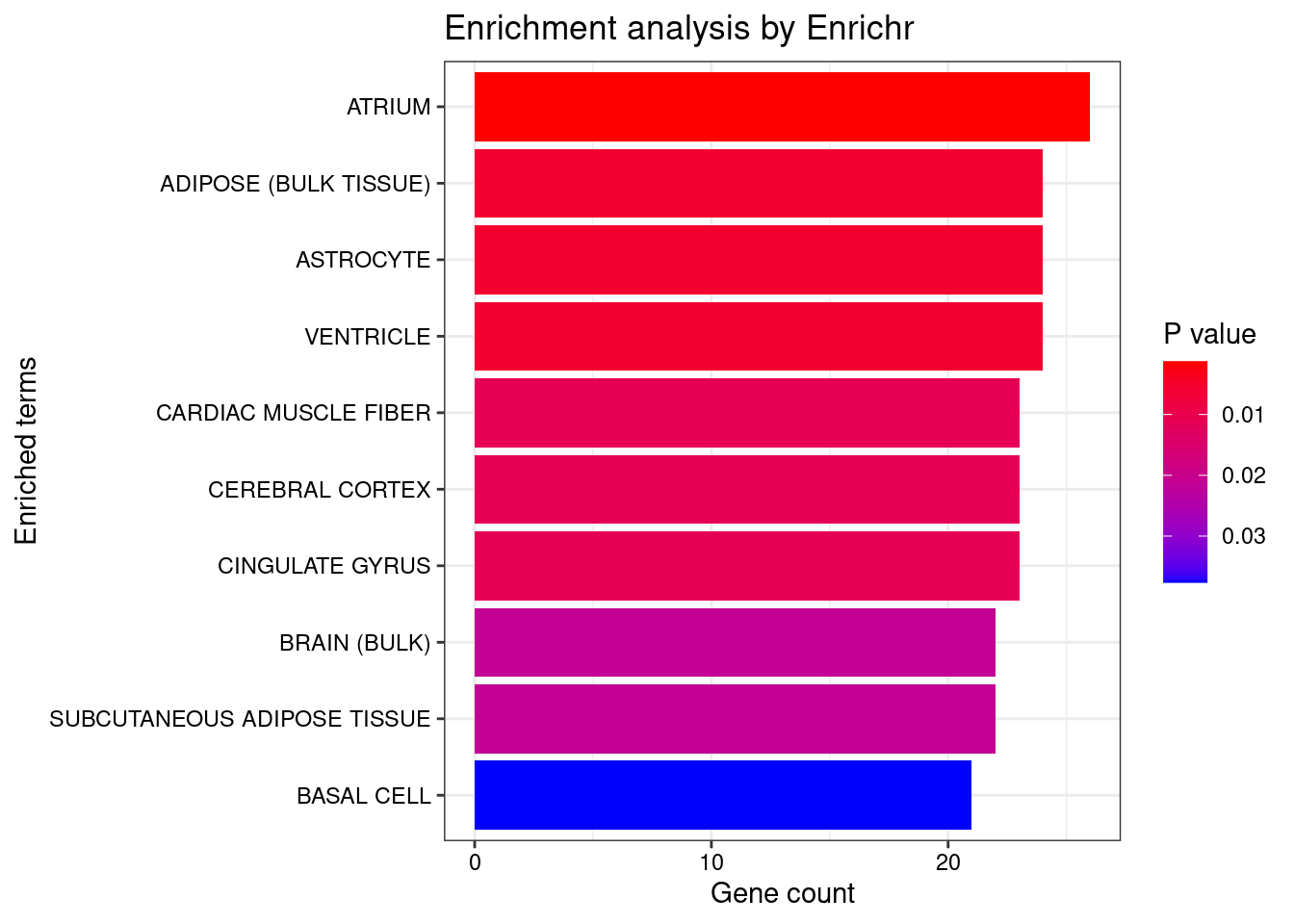
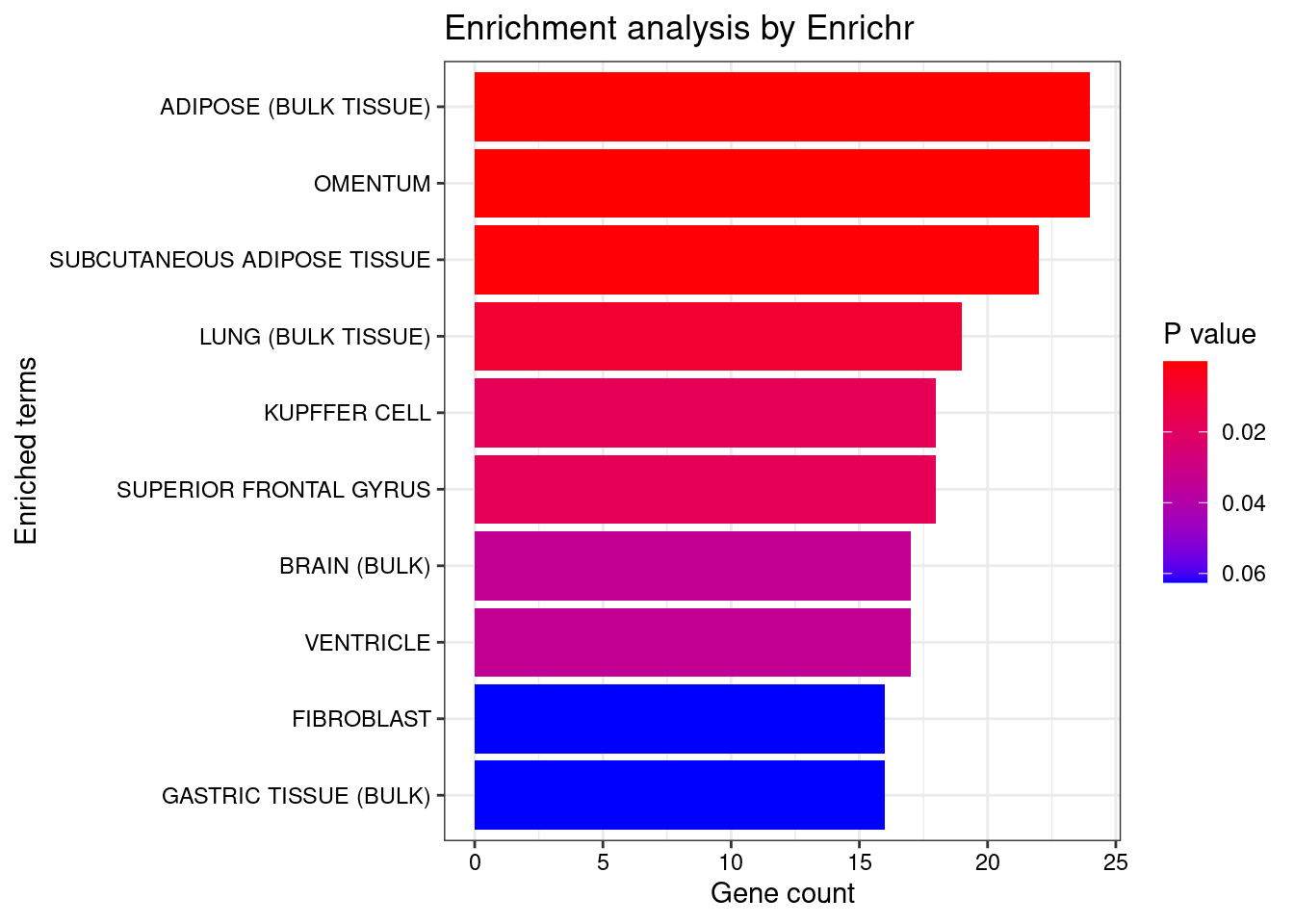
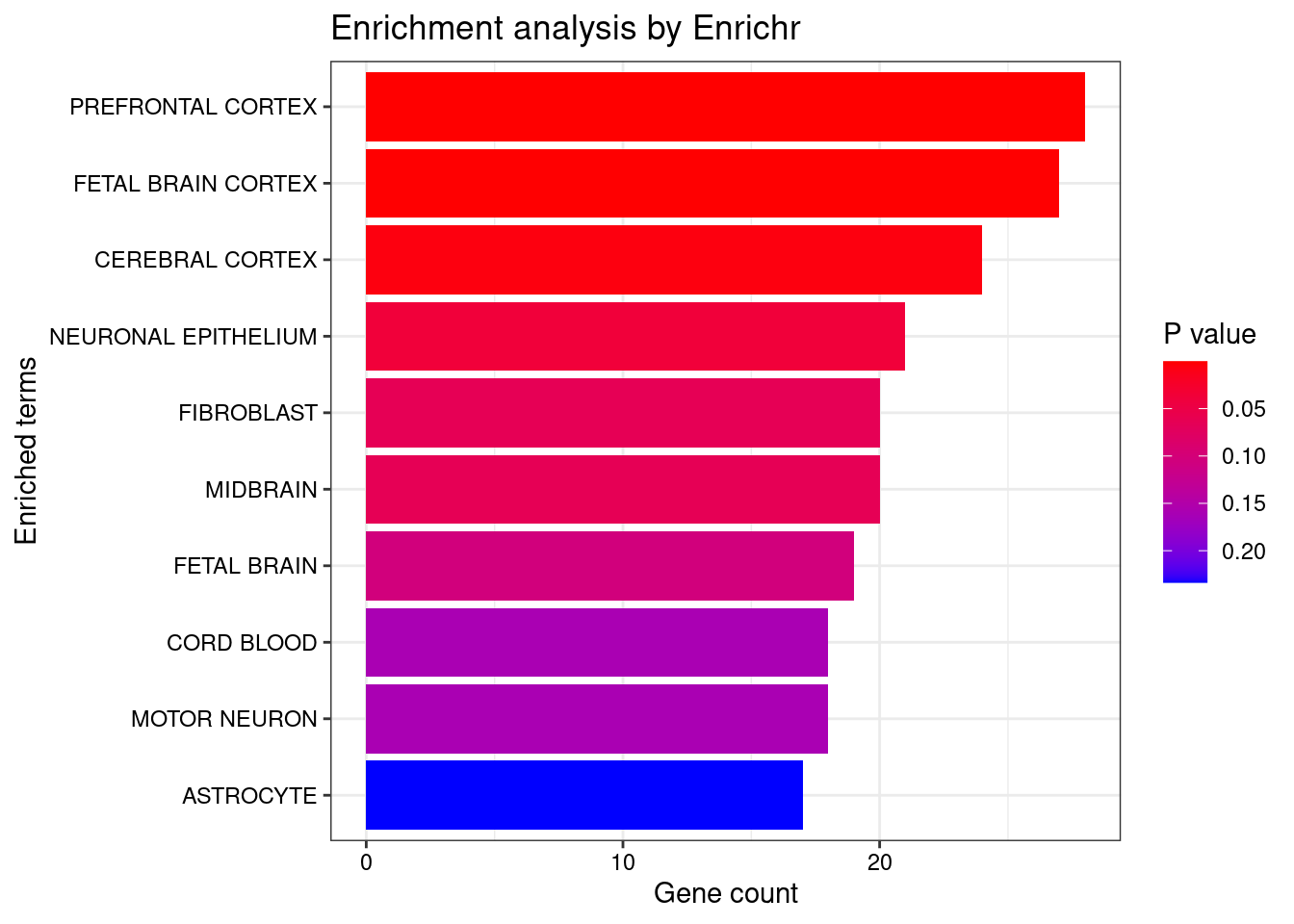
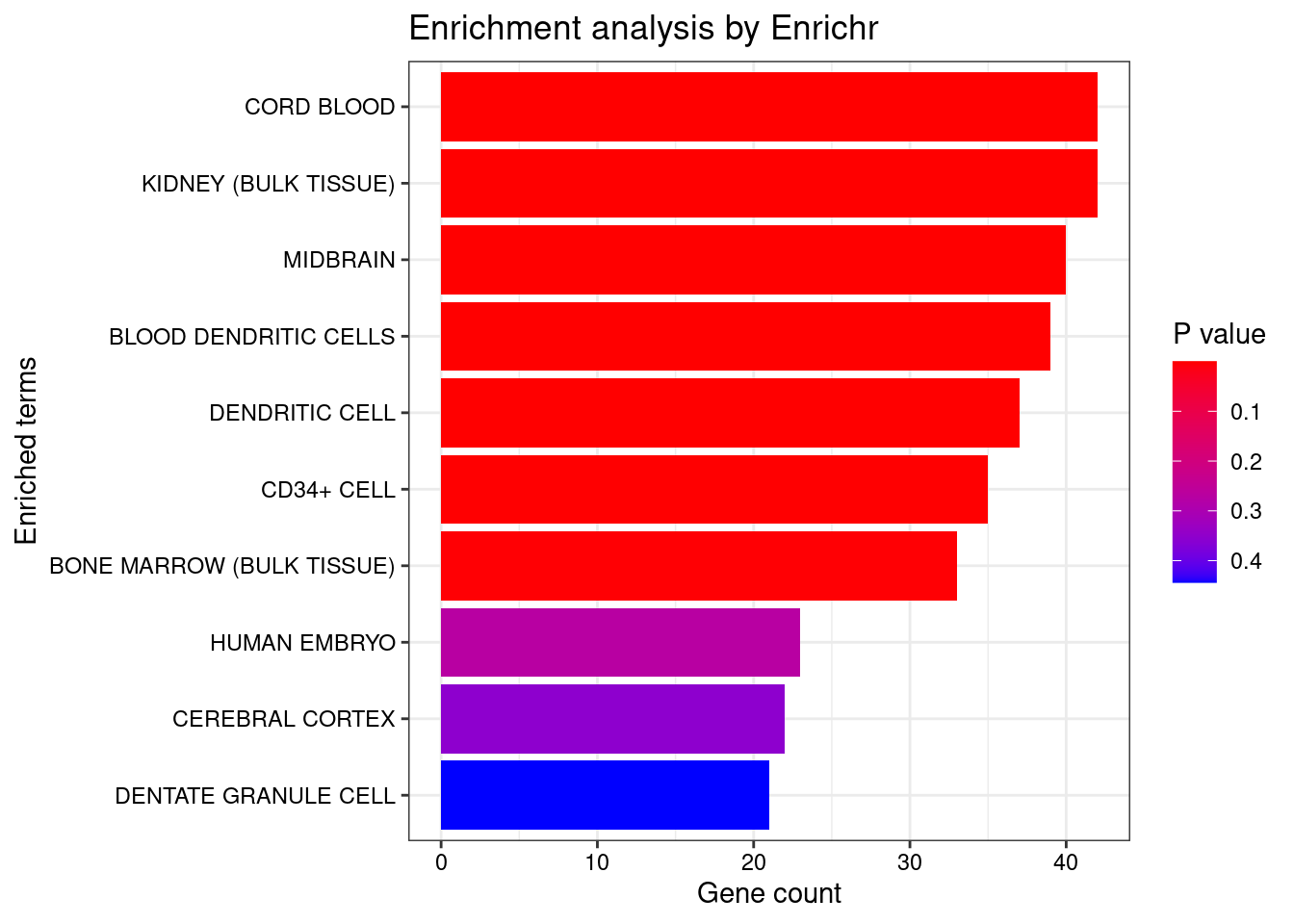
<- enrichr (genes.to.test.Cotan, dbs)
Uploading data to Enrichr... Done.
Querying ARCHS4_Tissues... Done.
Parsing results... Done.
plotEnrich (enriched[[1 ]], showTerms = 10 , numChar = 40 , y = "Count" , orderBy = "P.value" )
set.seed (123 )<- data.frame (Terms = str_split (enriched[[1 ]]$ Term[1 : 20 ],pattern = " CL" ,simplify = T )[,1 ],Scores = enriched[[1 ]]$ Combined.Score[1 : 20 ])wordcloud (wordcloud_data$ Terms, wordcloud_data$ Scores, scale = c (3 , 1 ), min.freq = 1 , random.order= FALSE , rot.per= 0.1 ,colors= brewer.pal (8 , "Dark2" ))
Cortical or hippocampal glutamatergic subclass
<- ggVennDiagram (markers.list[3 : 4 ])
<- markers.list$ ` Cortical or hippocampal glutamatergic Seurat ` [! markers.list$ ` Cortical or hippocampal glutamatergic Seurat ` %in% markers.list$ ` Cortical or hippocampal glutamatergic Cotan ` ]<- getNumOfExpressingCells (fb150Obj)[genes.to.test]<- as.data.frame (df)colnames (df) <- "CellNumber" rownames (df) <- NULL $ type <- "Only Seurat Genes" <- as.data.frame (getNumOfExpressingCells (fb150Obj)[sample (getGenes (fb150Obj), size = length (rownames (df)))])rownames (df.bk) <- NULL colnames (df.bk) <- "CellNumber" $ type <- "Whole dataset Genes" <- rbind (df,df.bk)library (plyr)library (scales) <- ddply (df, "type" , summarise, grp.mean= mean (CellNumber))ggplot (df,aes (x= CellNumber,fill= type))+ scale_fill_manual (values= c ("#E69F00" , "#56B4E9" ))+ geom_histogram (aes (y= ..density..), position= "identity" , alpha= 0.5 ,bins = 50 )+ geom_vline (data= mu, aes (xintercept= grp.mean, color= type),linetype= "dashed" )+ geom_density (alpha= 0.6 )+ scale_y_continuous (labels = percent, name = "percent" ) + theme_classic ()+ theme (legend.position= "bottom" )
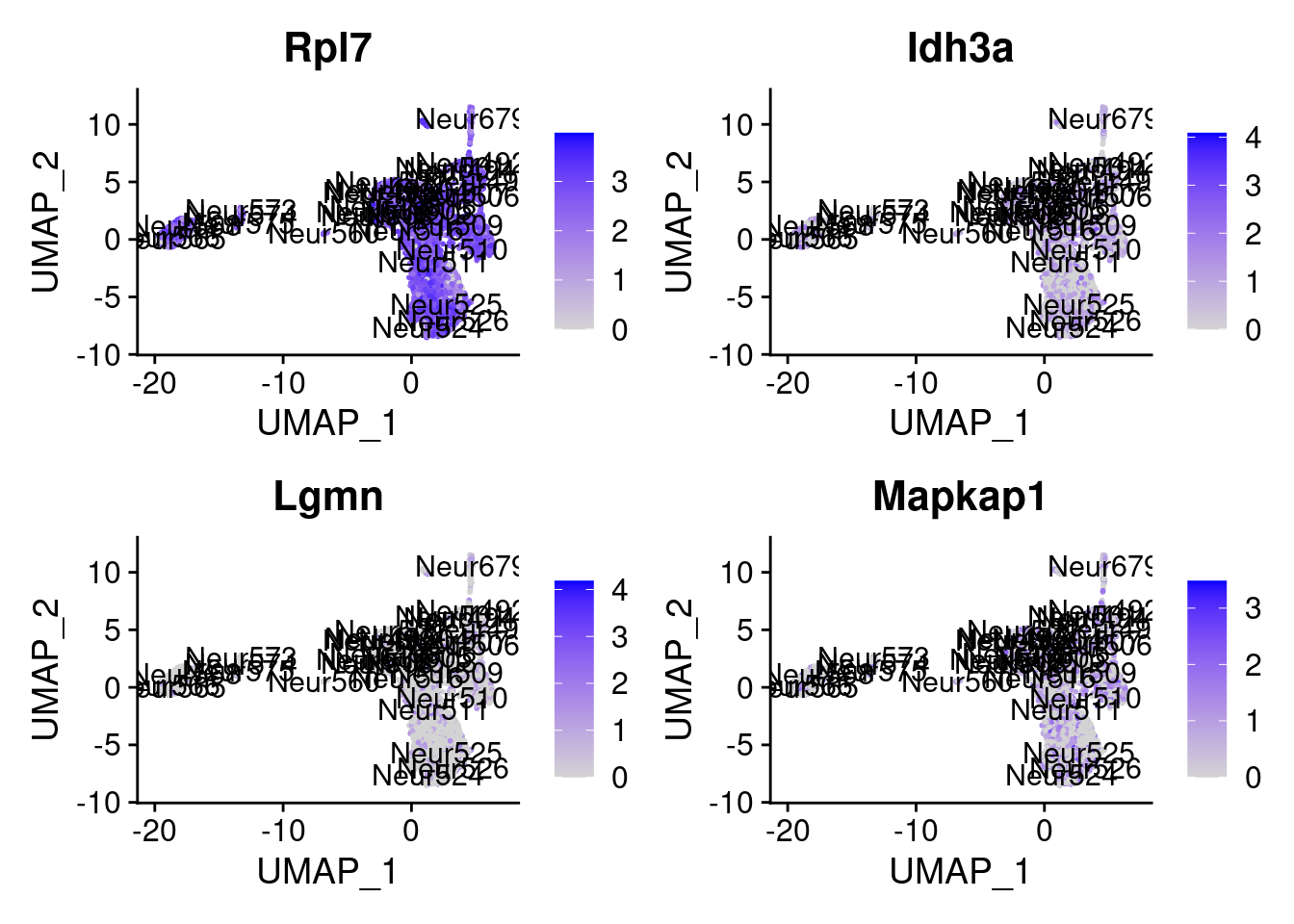
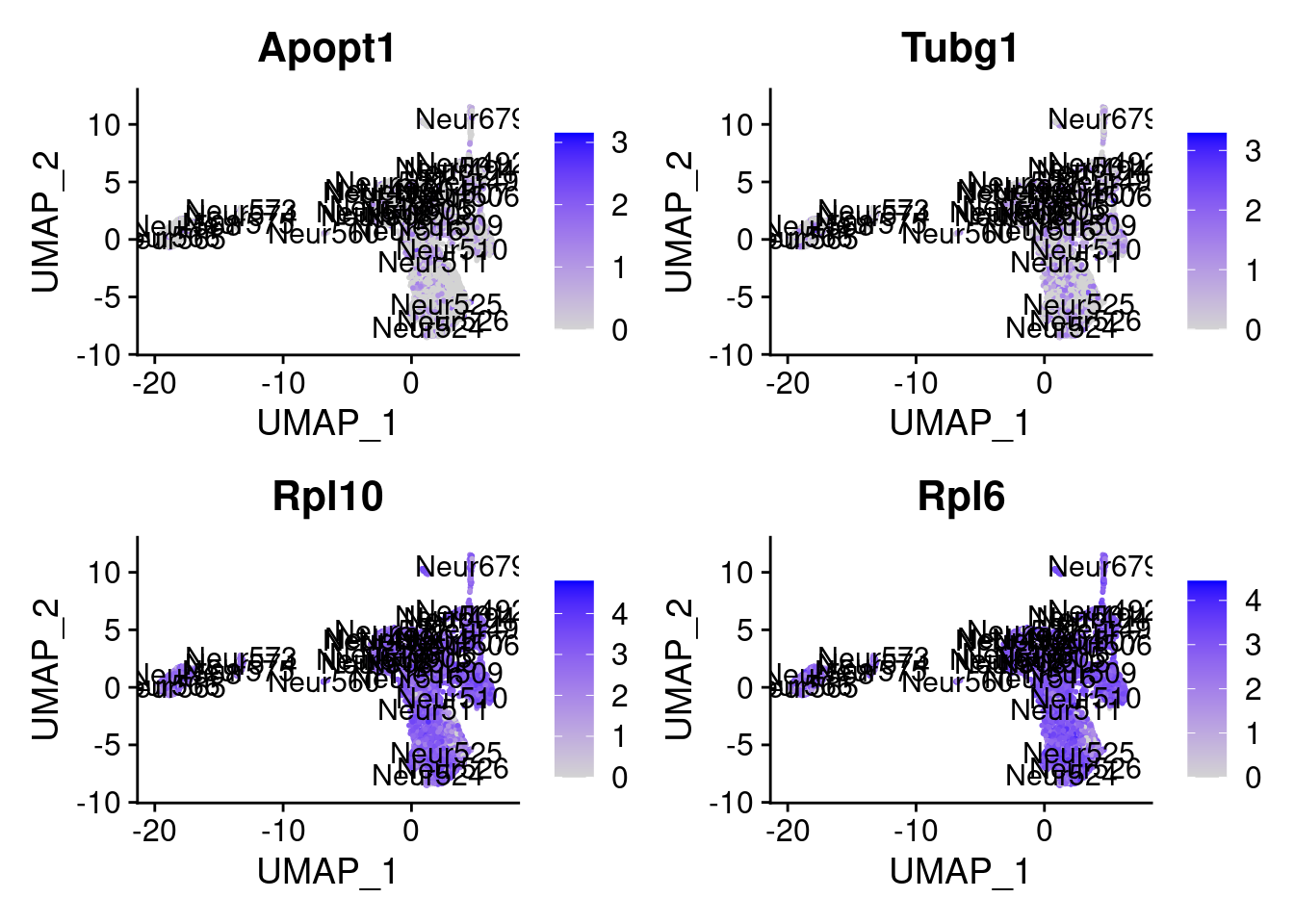
set.seed (111 )<- sample (genes.to.test,size = 18 )
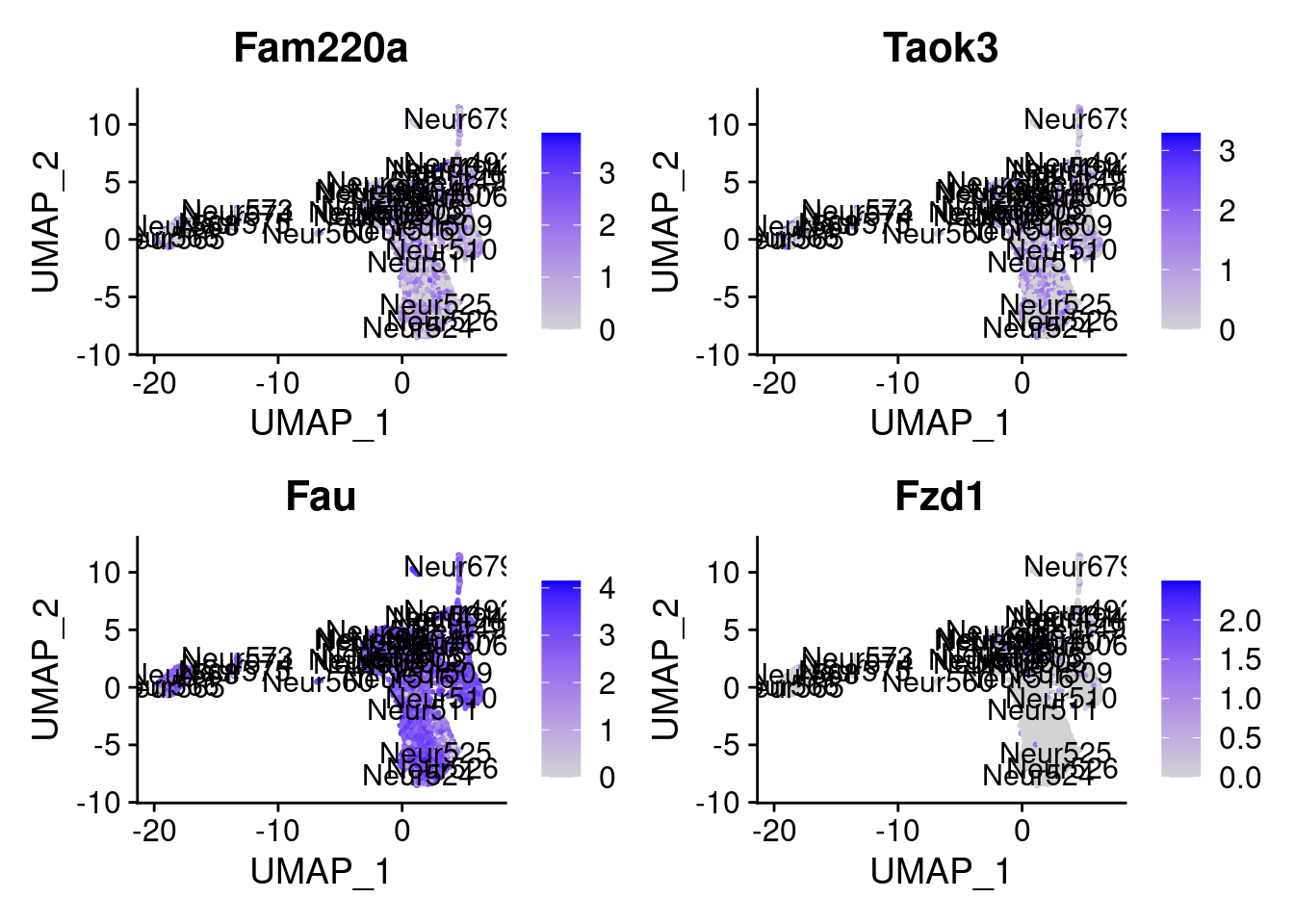
[1] "Fam220a" "Taok3" "Fau" "Fzd1" "Rpl7" "Idh3a" "Lgmn"
[8] "Mapkap1" "Apopt1" "Tubg1" "Rpl10" "Rpl6" "Ankrd45" "Nsa2"
[15] "Reep2" "Rpl14" "Egfem1" "Letm1"
= 0 for (g in c (0 : 2 )) {= g* 4 plot (FeaturePlot (seurat.obj,features = genes[n+ c (1 : 4 )], label = T))
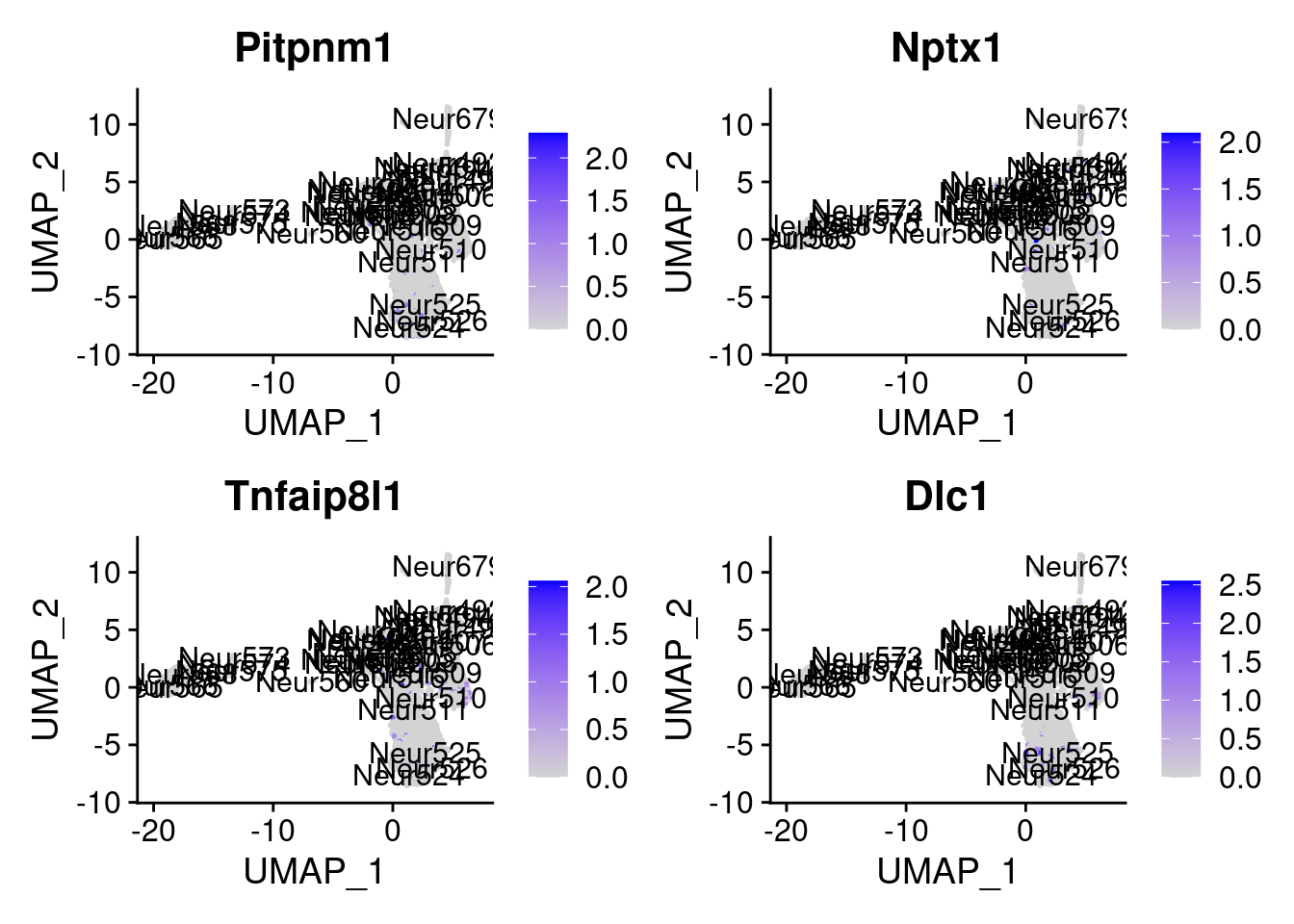
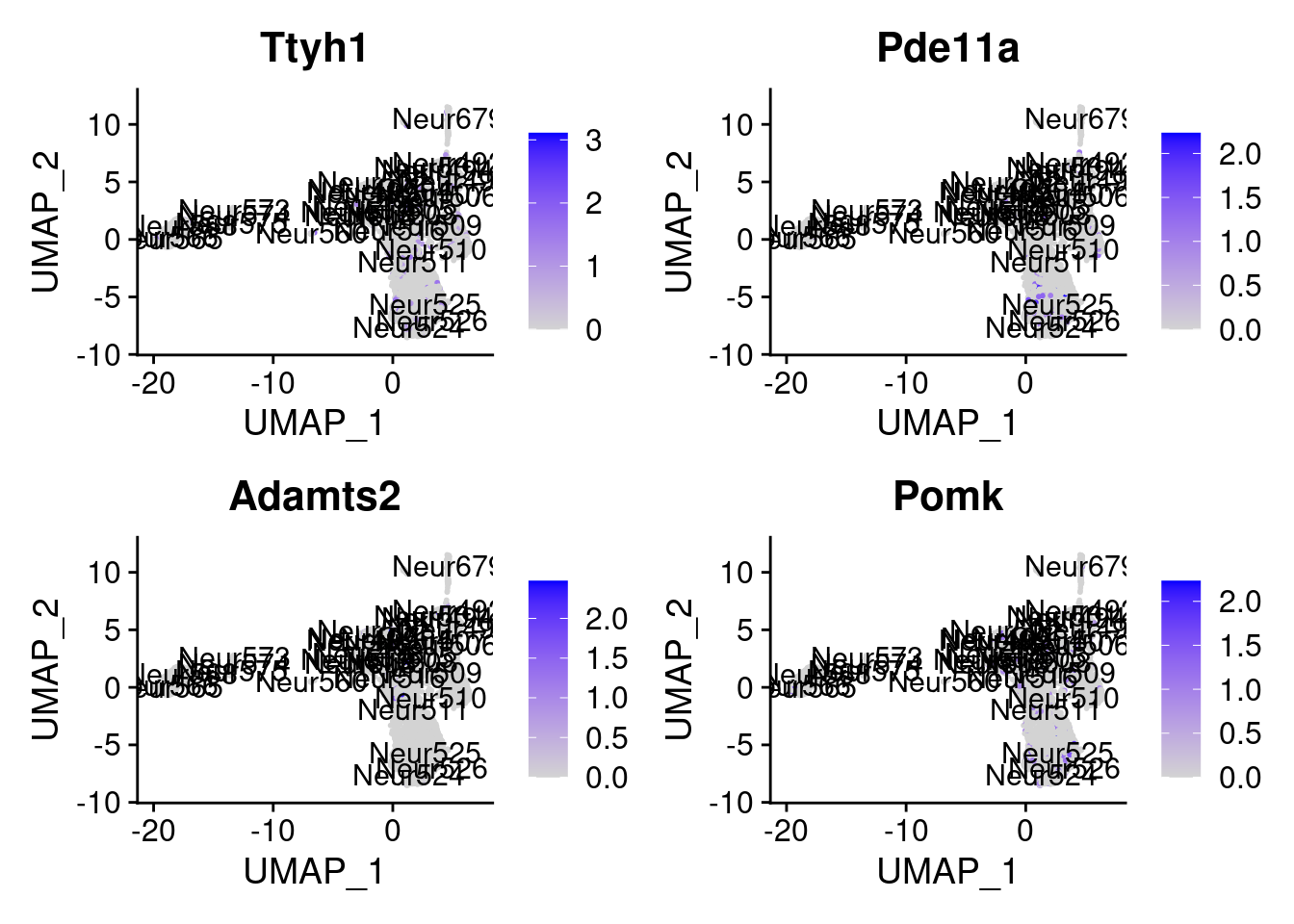
<- markers.list$ ` Cortical or hippocampal glutamatergic Cotan ` [! markers.list$ ` Cortical or hippocampal glutamatergic Cotan ` %in% markers.list$ ` Cortical or hippocampal glutamatergic Seurat ` ]set.seed (11 )<- sample (genes.to.test.Cotan,size = 30 )
[1] "Pitpnm1" "Nptx1" "Tnfaip8l1" "Dlc1"
[5] "Ttyh1" "Pde11a" "Adamts2" "Pomk"
[9] "D6Ertd474e" "Car12" "Rspo3" "Gm43517"
[13] "X3830406C13Rik" "Grp" "E2f1" "Ntf3"
[17] "Stim2" "D030068K23Rik" "Tspan17" "Ube2t"
[21] "Impa2" "Egfl6" "Serping1" "Pttg1"
[25] "Sh3bgrl3" "Mcrip2" "Map1lc3a" "Nrn1"
[29] "Gm42997" "AC124490.1"
= 0 for (g in c (0 : 2 )) {= g* 4 plot (FeaturePlot (seurat.obj,features = genes[n+ c (1 : 4 )], label = T))
<- getNumOfExpressingCells (fb150Obj)[genes.to.test.Cotan]<- as.data.frame (df)colnames (df) <- "CellNumber" rownames (df) <- NULL $ type <- "Only Cotan Genes" <- as.data.frame (getNumOfExpressingCells (fb150Obj)[sample (getGenes (fb150Obj), size = length (rownames (df)))])rownames (df.bk) <- NULL colnames (df.bk) <- "CellNumber" $ type <- "Whole dataset Genes" <- rbind (df,df.bk)<- ddply (df, "type" , summarise, grp.mean= mean (CellNumber))ggplot (df,aes (x= CellNumber,fill= type))+ scale_fill_manual (values= c ("#C69AFF" , "#56B4E9" ))+ geom_histogram (aes (y= ..density..), position= "identity" , alpha= 0.5 ,bins = 25 )+ xlim (0 ,4800 )+ geom_vline (data= mu, aes (xintercept= grp.mean, color= type),linetype= "dashed" )+ geom_density (alpha= 0.6 )+ scale_y_continuous (labels = percent, name = "percent" ) + theme_classic ()+ theme (legend.position= "bottom" )
<- seurat.obj.markers.Subclass[seurat.obj.markers.Subclass$ cluster == "Cortical or hippocampal glutamatergic" & seurat.obj.markers.Subclass$ gene %in% genes.to.test,]$ gene[1 : length (genes.to.test.Cotan)]<- enrichr (genes.to.testTop, dbs)
Uploading data to Enrichr... Done.
Querying ARCHS4_Tissues... Done.
Parsing results... Done.
plotEnrich (enriched[[1 ]], showTerms = 10 , numChar = 40 , y = "Count" , orderBy = "P.value" )
set.seed (12333 )<- data.frame (Terms = str_split (enriched[[1 ]]$ Term[1 : 8 ],pattern = " CL" ,simplify = T )[,1 ],Scores = enriched[[1 ]]$ Combined.Score[1 : 8 ])wordcloud (wordcloud_data$ Terms, wordcloud_data$ Scores, scale = c (2 , 0.5 ), min.freq = 1 , random.order= FALSE , rot.per= 0.1 ,colors= brewer.pal (8 , "Dark2" ))
<- enrichr (genes.to.test.Cotan, dbs)
Uploading data to Enrichr... Done.
Querying ARCHS4_Tissues... Done.
Parsing results... Done.
plotEnrich (enriched[[1 ]], showTerms = 10 , numChar = 40 , y = "Count" , orderBy = "P.value" )
set.seed (1233 )<- data.frame (Terms = str_split (enriched[[1 ]]$ Term[1 : 20 ],pattern = " CL" ,simplify = T )[,1 ],Scores = enriched[[1 ]]$ Combined.Score[1 : 20 ])wordcloud (wordcloud_data$ Terms, wordcloud_data$ Scores, scale = c (3 , 1 ), min.freq = 1 , random.order= FALSE , rot.per= 0.1 ,colors= brewer.pal (8 , "Dark2" ))
Forebrain GABAergic subclass
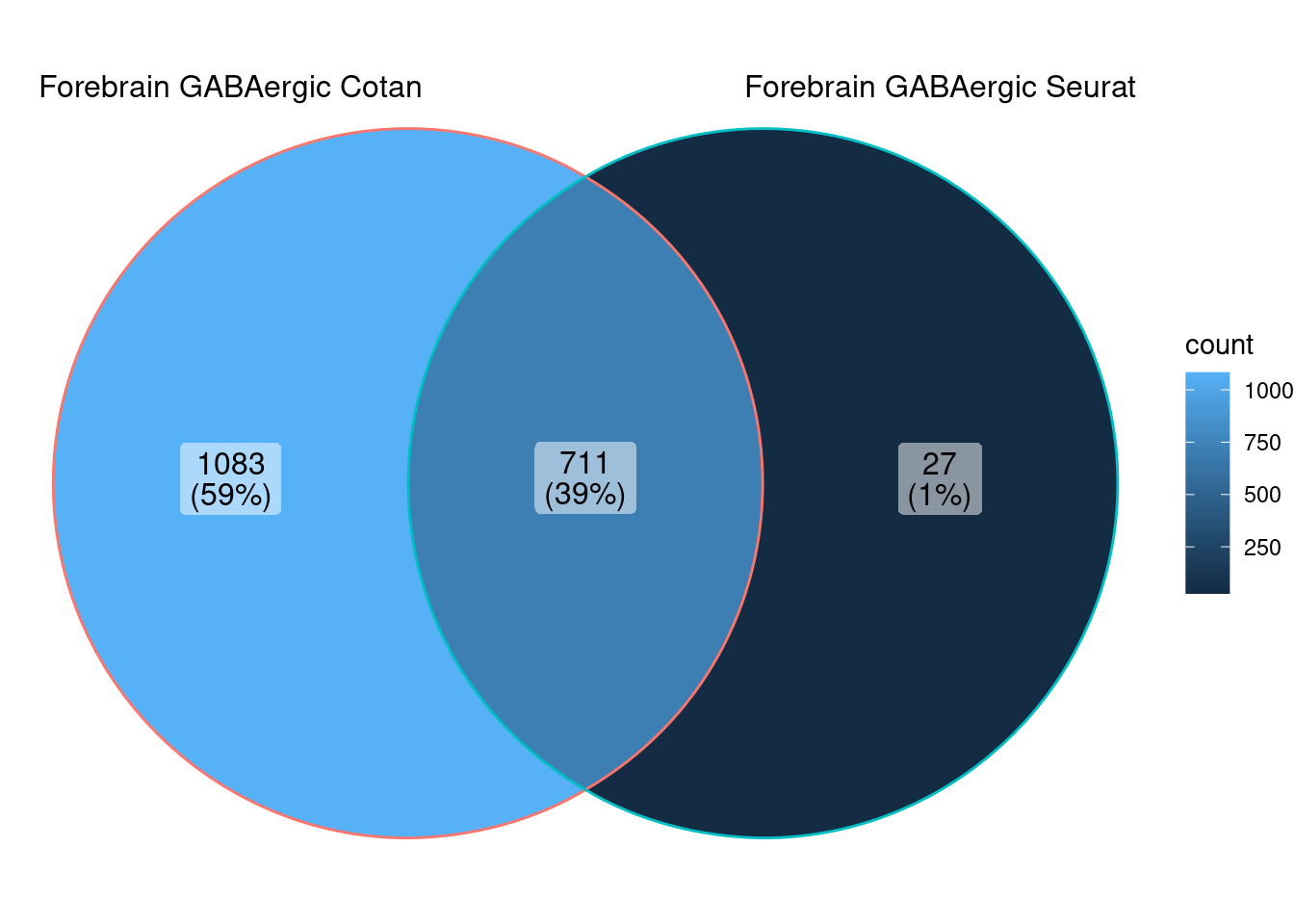
<- ggVennDiagram (markers.list[5 : 6 ])
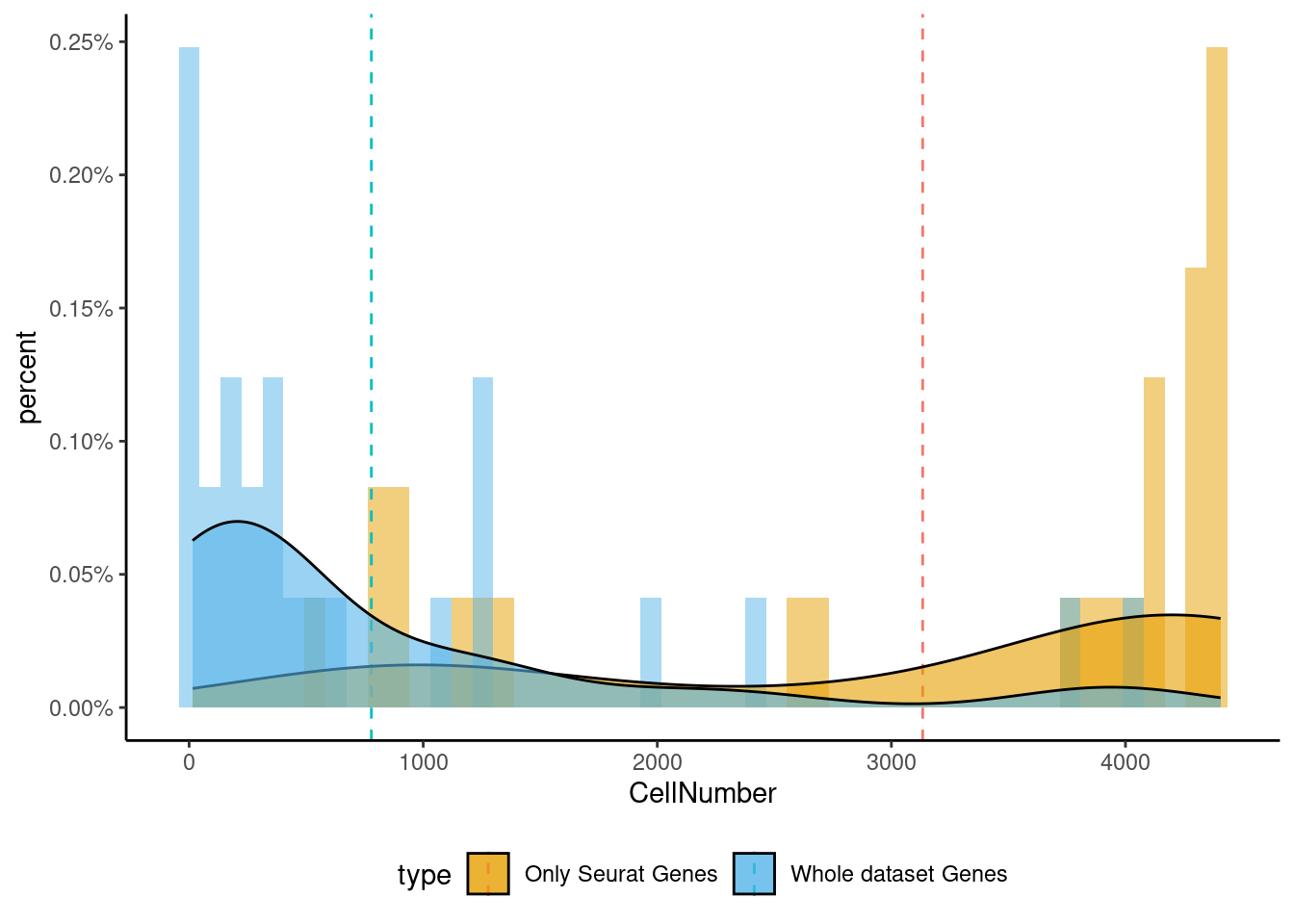
<- markers.list$ ` Forebrain GABAergic Seurat ` [! markers.list$ ` Forebrain GABAergic Seurat ` %in% markers.list$ ` Forebrain GABAergic Cotan ` ]<- getNumOfExpressingCells (fb150Obj)[genes.to.test]<- as.data.frame (df)colnames (df) <- "CellNumber" rownames (df) <- NULL $ type <- "Only Seurat Genes" <- as.data.frame (getNumOfExpressingCells (fb150Obj)[sample (getGenes (fb150Obj), size = length (rownames (df)))])rownames (df.bk) <- NULL colnames (df.bk) <- "CellNumber" $ type <- "Whole dataset Genes" <- rbind (df,df.bk)library (plyr)library (scales) <- ddply (df, "type" , summarise, grp.mean= mean (CellNumber))ggplot (df,aes (x= CellNumber,fill= type))+ scale_fill_manual (values= c ("#E69F00" , "#56B4E9" ))+ geom_histogram (aes (y= ..density..), position= "identity" , alpha= 0.5 ,bins = 50 )+ geom_vline (data= mu, aes (xintercept= grp.mean, color= type),linetype= "dashed" )+ geom_density (alpha= 0.6 )+ scale_y_continuous (labels = percent, name = "percent" ) + theme_classic ()+ theme (legend.position= "bottom" )
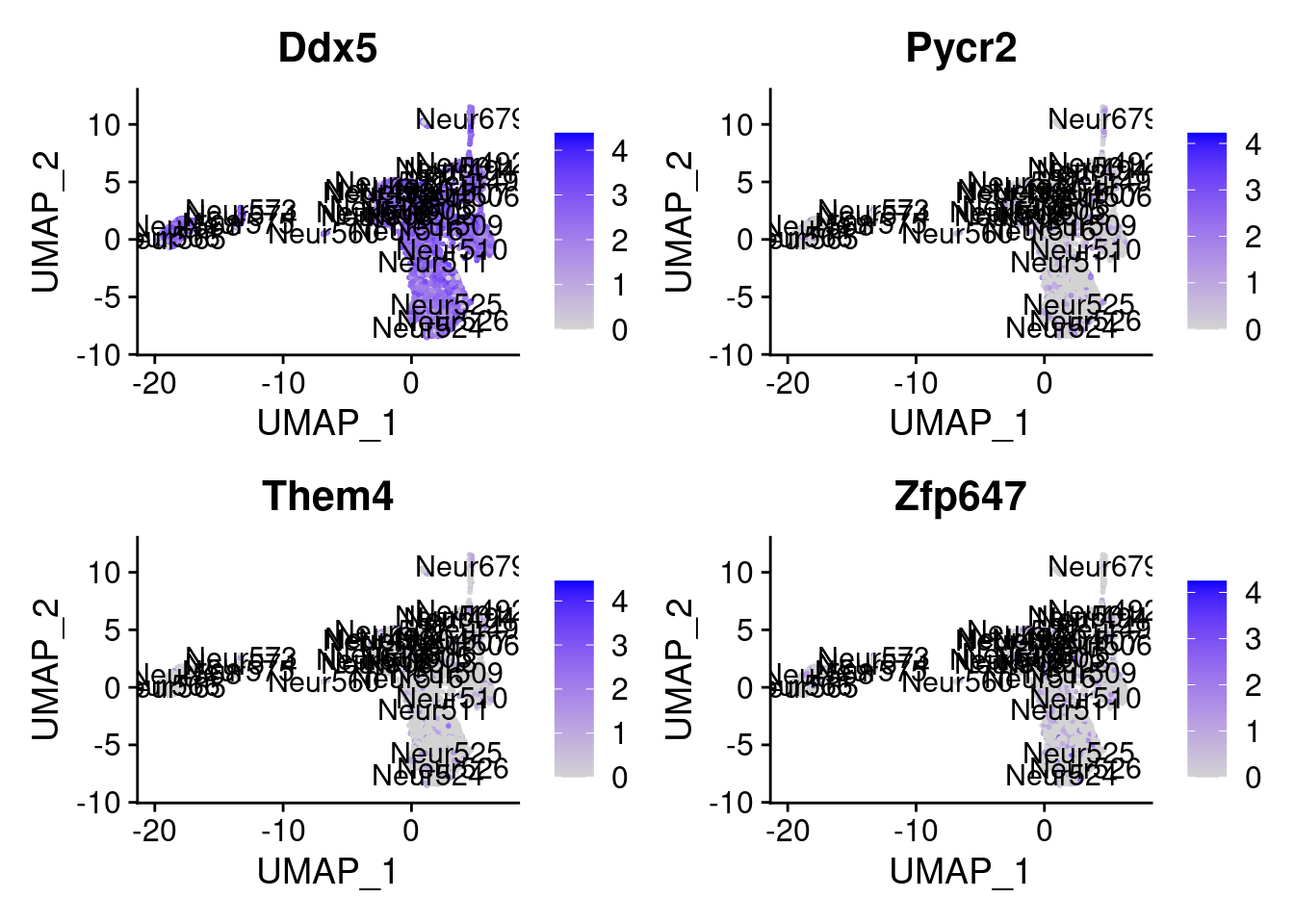
set.seed (111 )<- sample (genes.to.test,size = 18 )
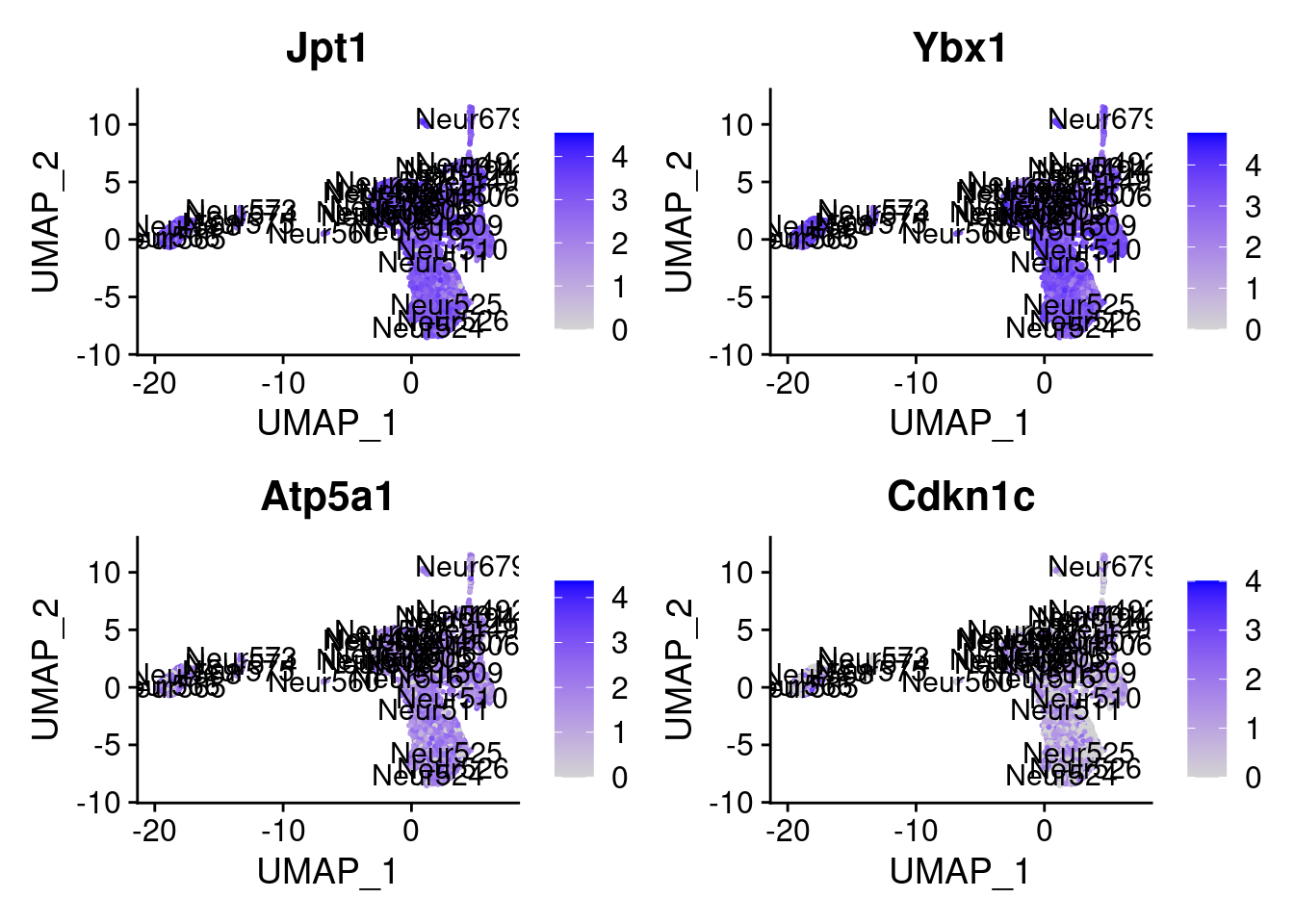
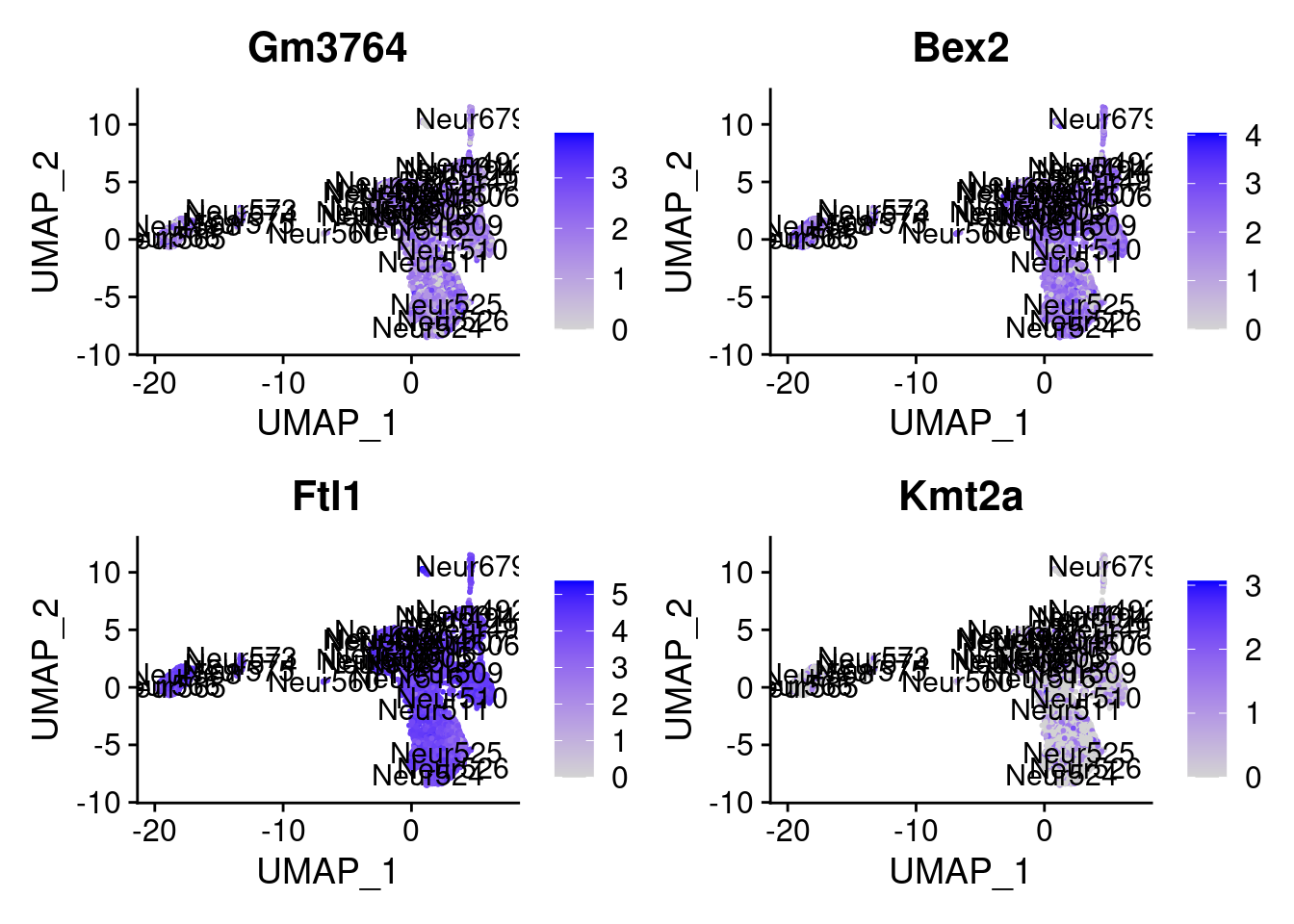
[1] "Ddx5" "Pycr2" "Them4" "Zfp647" "Jpt1" "Ybx1" "Atp5a1"
[8] "Cdkn1c" "Gm3764" "Bex2" "Ftl1" "Kmt2a" "Map2" "Cntnap2"
[15] "Pogk" "Rtn1" "Scg5" "H3f3b"
= 0 for (g in c (0 : 2 )) {= g* 4 plot (FeaturePlot (seurat.obj,features = genes[n+ c (1 : 4 )], label = T))
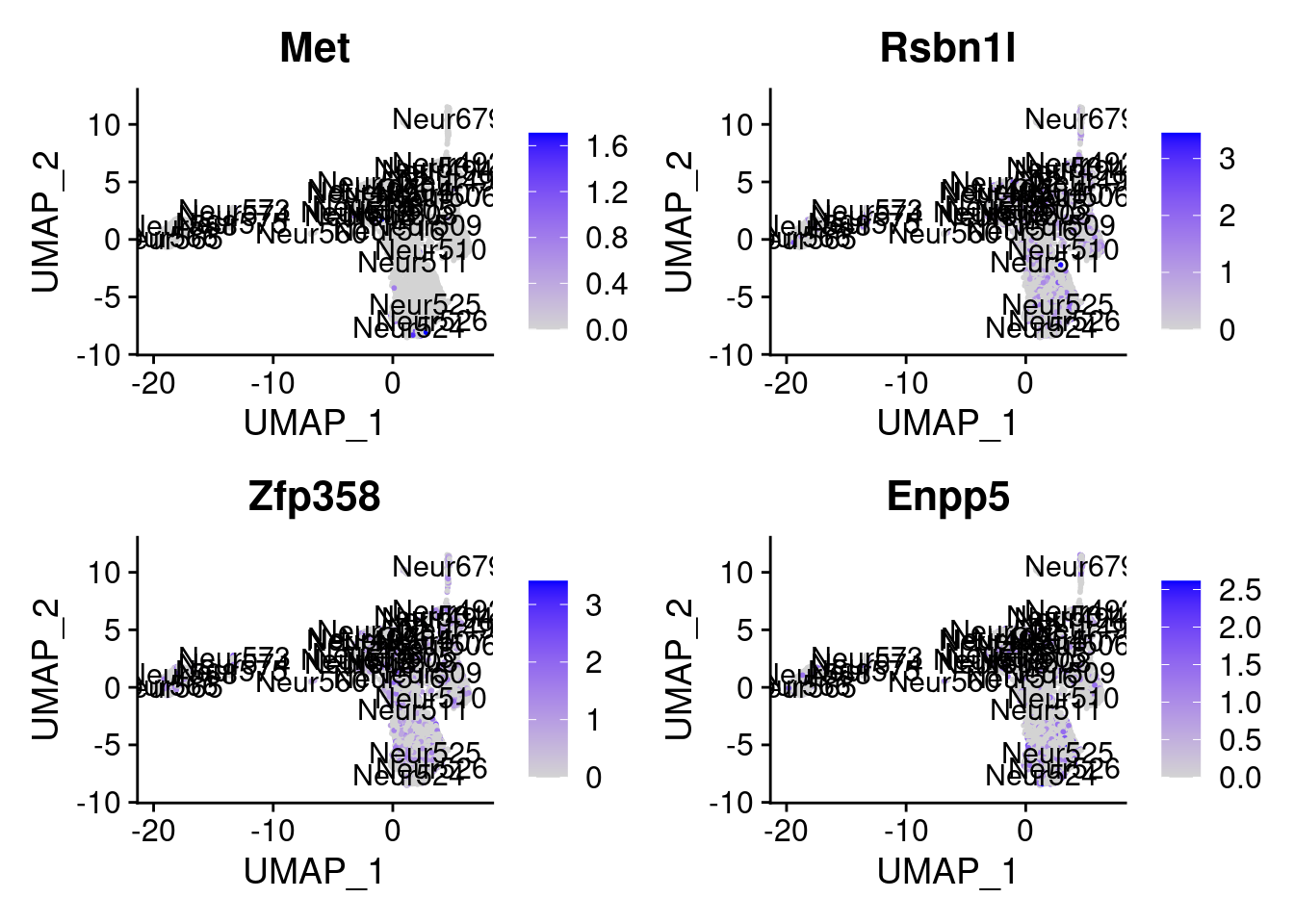
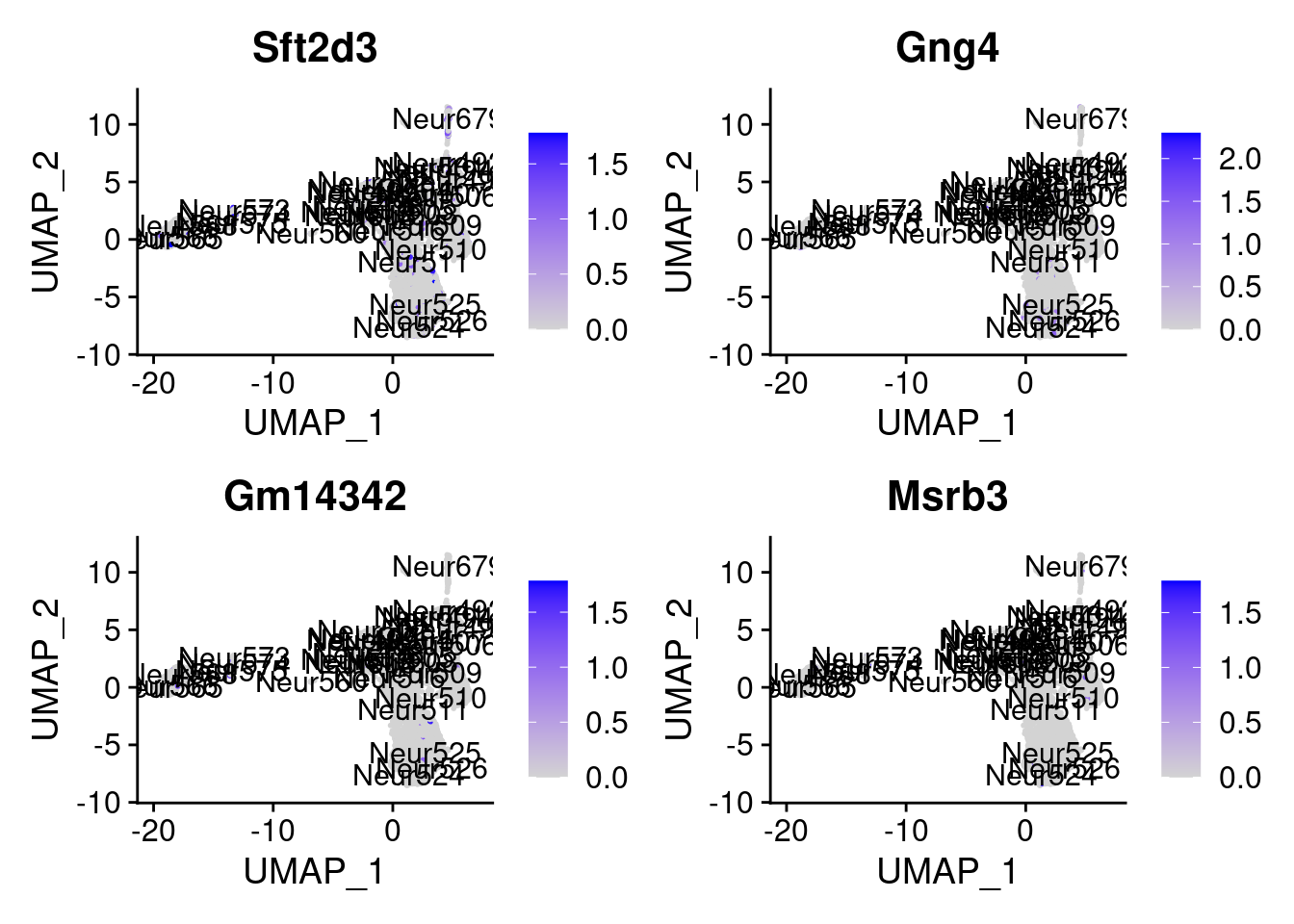
<- markers.list$ ` Forebrain GABAergic Cotan ` [! markers.list$ ` Forebrain GABAergic Cotan ` %in% markers.list$ ` Forebrain GABAergic Seurat ` ]set.seed (11 )<- sample (genes.to.test.Cotan,size = 30 )
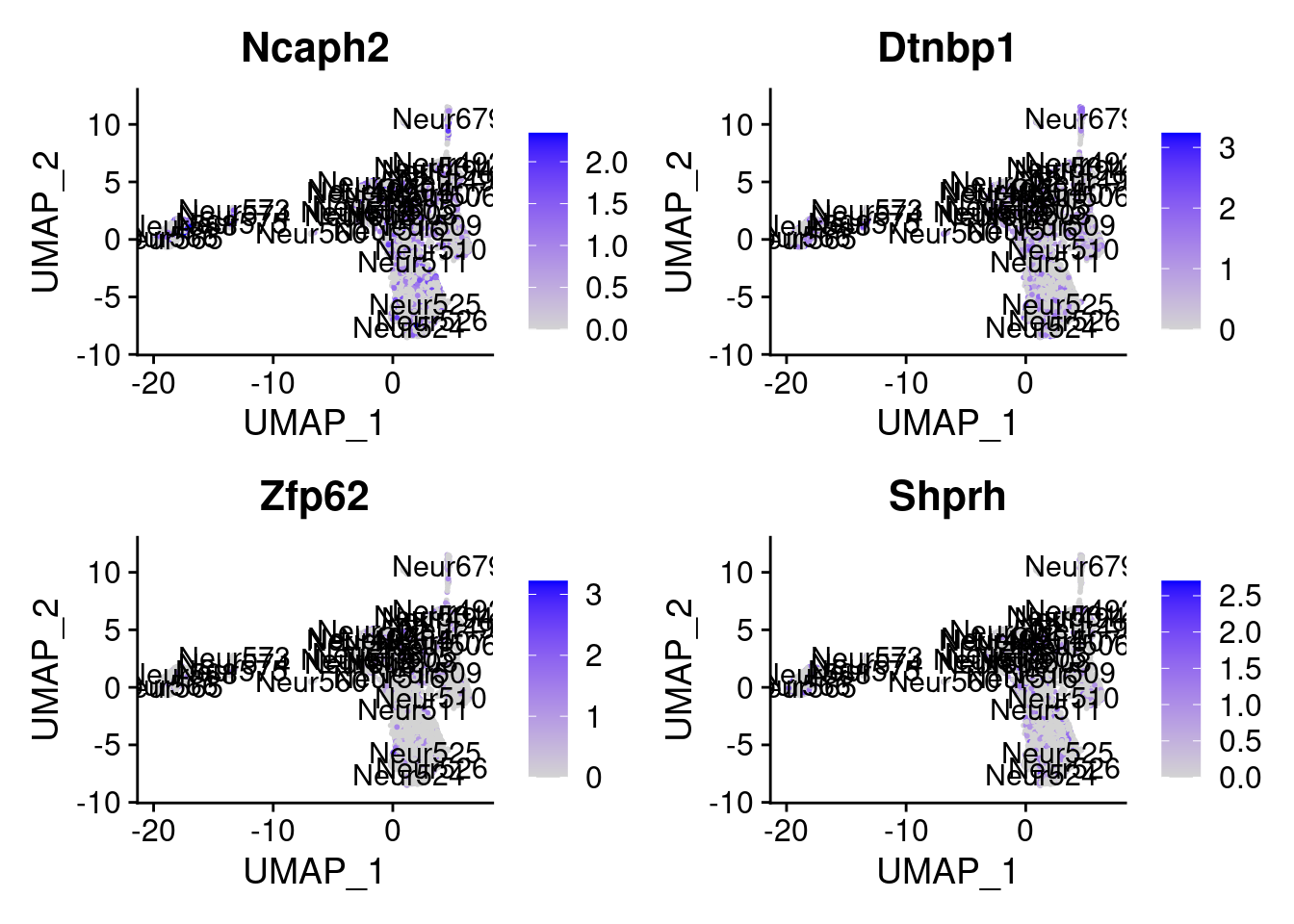
[1] "Met" "Rsbn1l" "Zfp358" "Enpp5" "Sft2d3"
[6] "Gng4" "Gm14342" "Msrb3" "Ncaph2" "Dtnbp1"
[11] "Zfp62" "Shprh" "Bmf" "AC174678.1" "Map7d1"
[16] "Srsf1" "Smim7" "Rab39b" "Depdc7" "Prkg1"
[21] "Rad21" "Brinp1" "Rbm5" "Laptm4b" "Dcbld2"
[26] "Slf2" "Fam173a" "Sh3bp5l" "Slc1a2" "Sstr1"
= 0 for (g in c (0 : 2 )) {= g* 4 plot (FeaturePlot (seurat.obj,features = genes[n+ c (1 : 4 )], label = T))
<- getNumOfExpressingCells (fb150Obj)[genes.to.test.Cotan]<- as.data.frame (df)colnames (df) <- "CellNumber" rownames (df) <- NULL $ type <- "Only Cotan Genes" <- as.data.frame (getNumOfExpressingCells (fb150Obj)[sample (getGenes (fb150Obj), size = length (rownames (df)))])rownames (df.bk) <- NULL colnames (df.bk) <- "CellNumber" $ type <- "Whole dataset Genes" <- rbind (df,df.bk)<- ddply (df, "type" , summarise, grp.mean= mean (CellNumber))ggplot (df,aes (x= CellNumber,fill= type))+ scale_fill_manual (values= c ("#C69AFF" , "#56B4E9" ))+ geom_histogram (aes (y= ..density..), position= "identity" , alpha= 0.5 ,bins = 25 )+ xlim (0 ,4800 )+ geom_vline (data= mu, aes (xintercept= grp.mean, color= type),linetype= "dashed" )+ geom_density (alpha= 0.6 )+ scale_y_continuous (labels = percent, name = "percent" ) + theme_classic ()+ theme (legend.position= "bottom" )
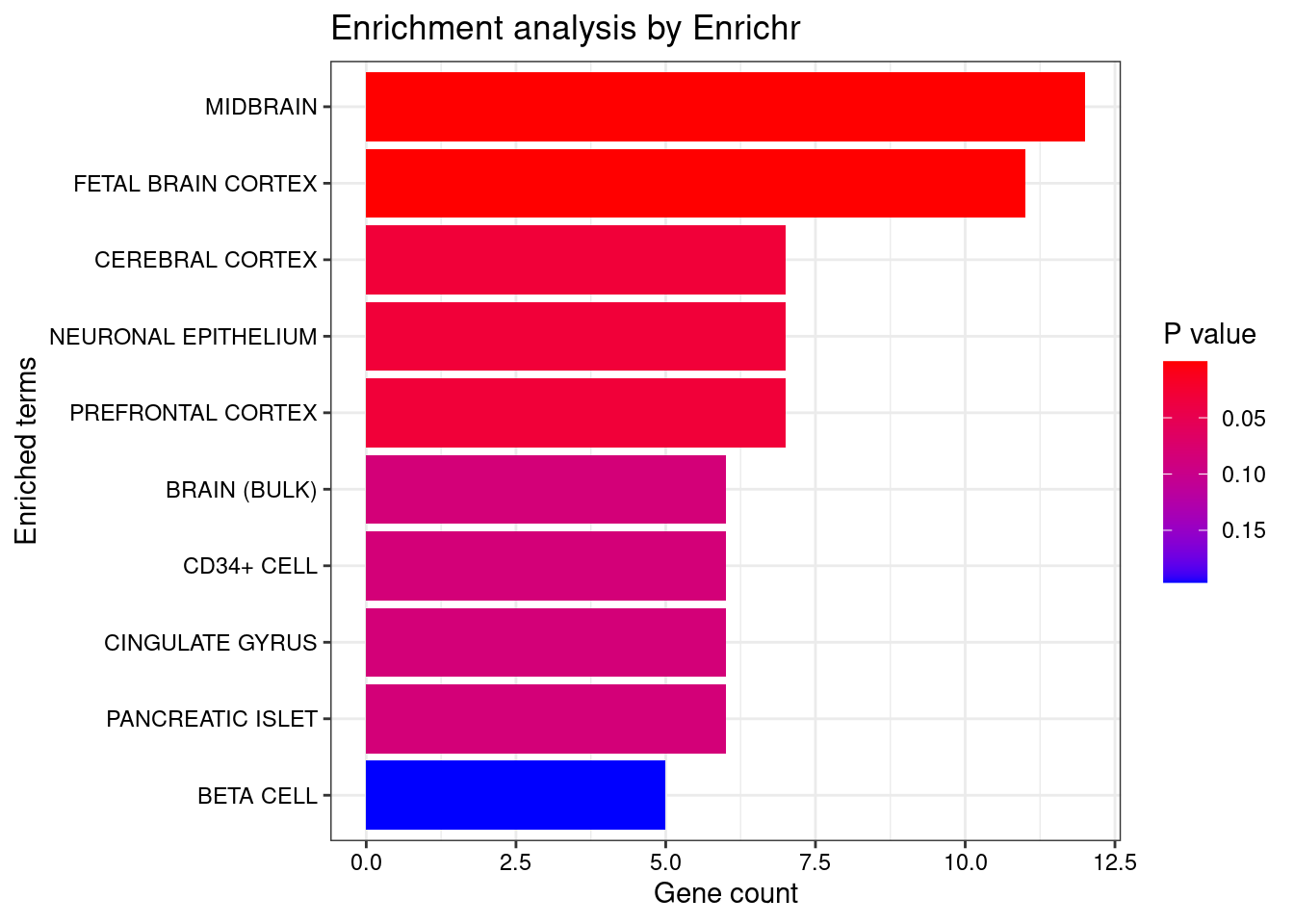
<- enrichr (genes.to.test, dbs)
Uploading data to Enrichr... Done.
Querying ARCHS4_Tissues... Done.
Parsing results... Done.
plotEnrich (enriched[[1 ]], showTerms = 10 , numChar = 40 , y = "Count" , orderBy = "P.value" )
set.seed (12333 )<- data.frame (Terms = str_split (enriched[[1 ]]$ Term[1 : 5 ],pattern = " CL" ,simplify = T )[,1 ],Scores = enriched[[1 ]]$ Combined.Score[1 : 5 ])wordcloud (wordcloud_data$ Terms, wordcloud_data$ Scores, scale = c (3 , 1 ), min.freq = 1 , random.order= FALSE , rot.per= 0.1 ,colors= brewer.pal (8 , "Dark2" ))
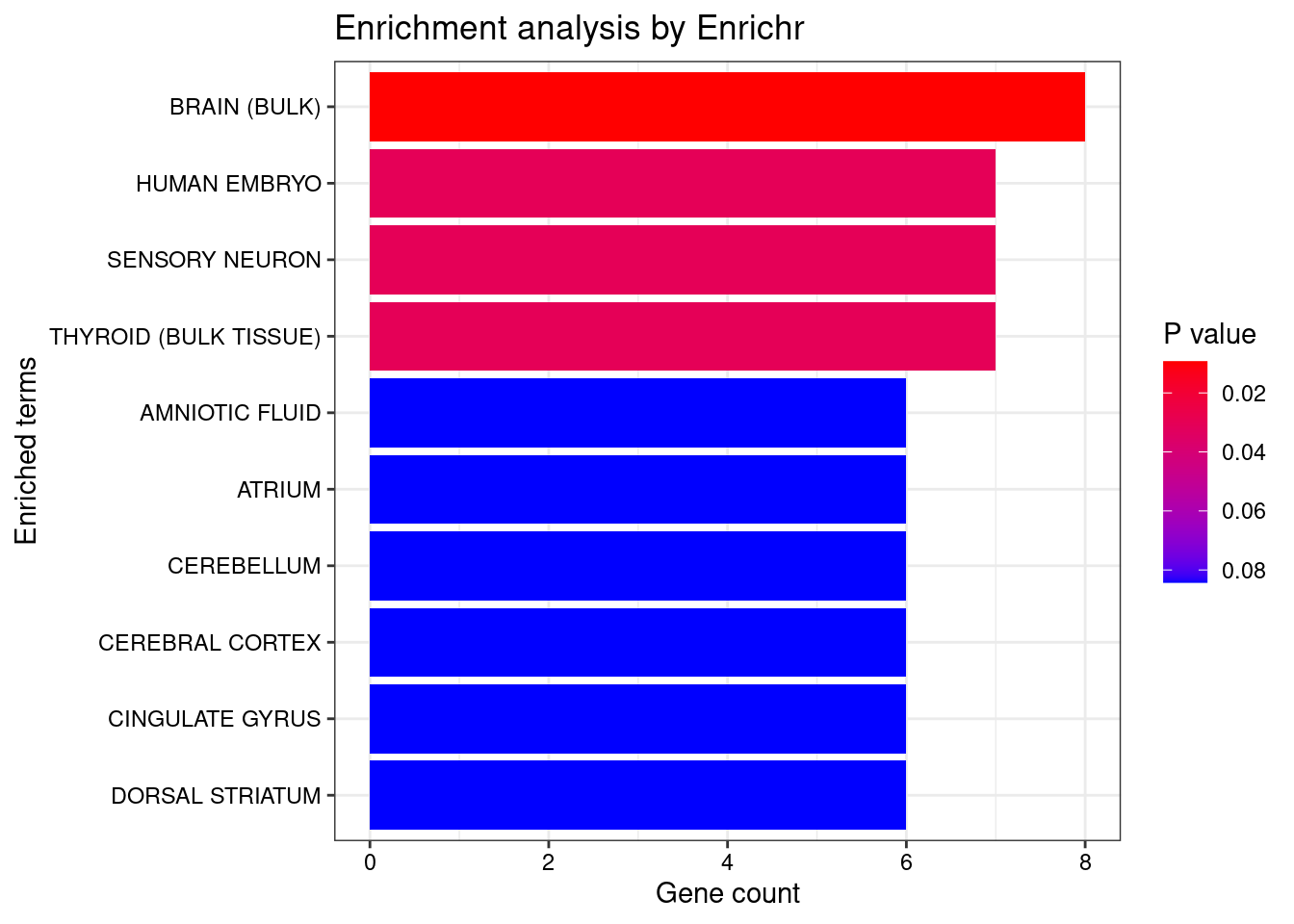
<- dea.Subclass$ ` p-value ` [rownames (dea.Subclass$ ` p-value ` ) %in% genes.to.test.Cotan,]<- rownames (subset.pval[order (subset.pval$ ` Forebrain GABAergic ` ,decreasing = F),])[1 : length (genes.to.test)]<- enrichr (genes.to.test.Cotan.Top, dbs)
Uploading data to Enrichr... Done.
Querying ARCHS4_Tissues... Done.
Parsing results... Done.
plotEnrich (enriched[[1 ]], showTerms = 10 , numChar = 40 , y = "Count" , orderBy = "P.value" )
set.seed (123 )<- data.frame (Terms = str_split (enriched[[1 ]]$ Term,pattern = " CL" ,simplify = T )[,1 ],Scores = enriched[[1 ]]$ Combined.Score)wordcloud (wordcloud_data$ Terms, wordcloud_data$ Scores, scale = c (3 , 1 ), min.freq = 1 , random.order= FALSE , rot.per= 0.1 ,colors= brewer.pal (8 , "Dark2" ))
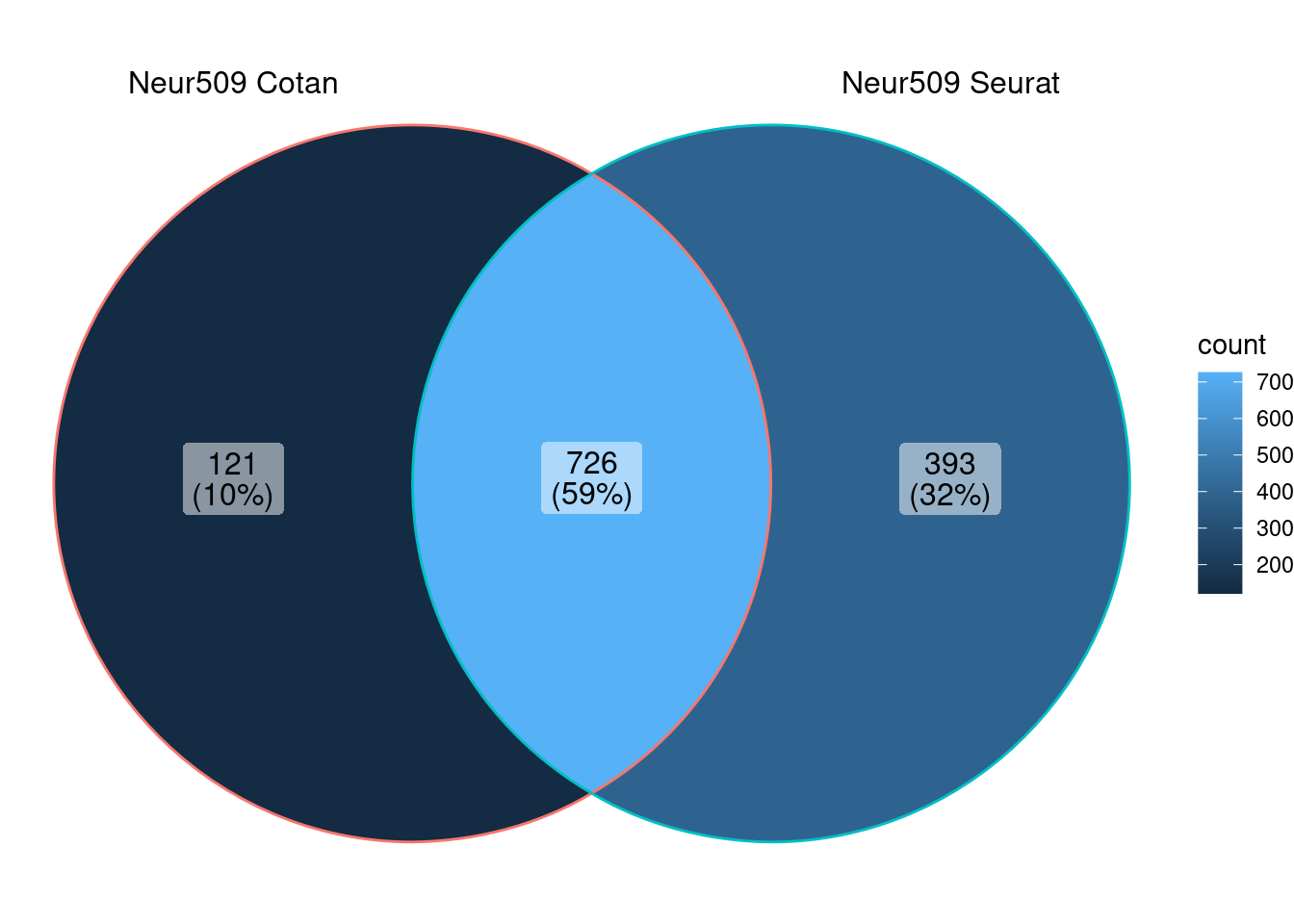
Cluster Neur509
# markers.list.namesNeur509 <- col_concat(crossing(colnames(dea.ClusterName$coex),c("Seurat","Cotan")),sep = " ") # # markers.listNeur509 <- vector("list", length(markers.list.namesNeur509)) # names(markers.listNeur509) <- markers.list.namesNeur509 <- list ("Neur509 Cotan" = NA ,"Neur509 Seurat" = NA )# I take positive coex significant genes for (cl.name in "Neur509" ) {<- rownames (dea.ClusterName$ coex[dea.ClusterName$ ` p-value ` [,cl.name] < 0.01 & dea.ClusterName$ coex[,cl.name] > 0 ,])paste0 (cl.name," Cotan" )]] <- genes#For seurat for (cl.name in "Neur509" ) {<- seurat.obj.markers.ClusterName[seurat.obj.markers.ClusterName$ cluster == cl.name & seurat.obj.markers.ClusterName$ p_val < 0.01 ,]$ genepaste0 (cl.name," Seurat" )]] <- genes
ggVennDiagram (markers.listNeur509)
We can observe that there is a good overlap among the detected markers.
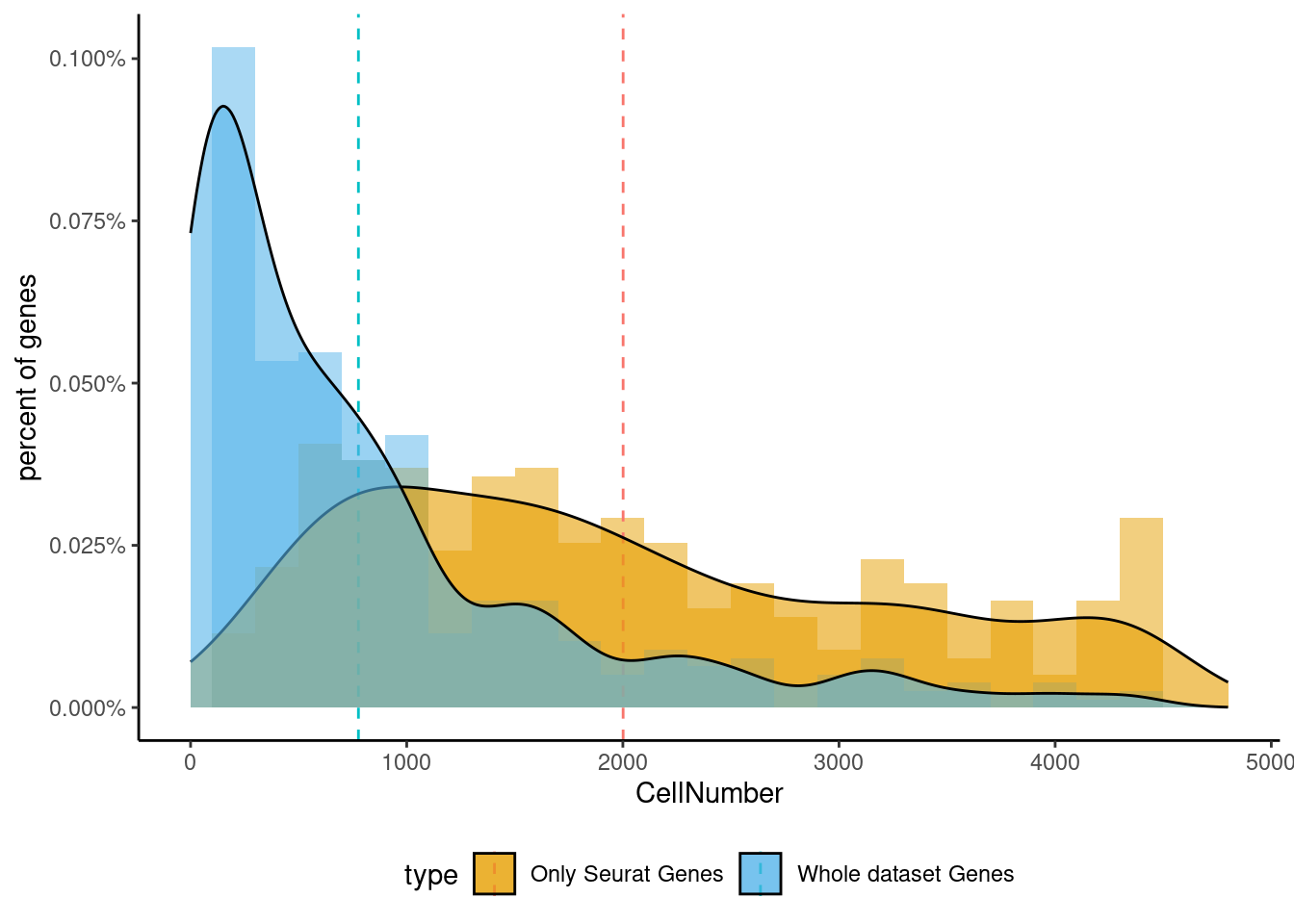
<- markers.listNeur509$ ` Neur509 Seurat ` [! markers.listNeur509$ ` Neur509 Seurat ` %in% markers.listNeur509$ ` Neur509 Cotan ` ]<- getNumOfExpressingCells (fb150Obj)[genes.to.test]<- as.data.frame (df)colnames (df) <- "CellNumber" rownames (df) <- NULL $ type <- "Only Seurat Genes" <- as.data.frame (getNumOfExpressingCells (fb150Obj)[sample (getGenes (fb150Obj), size = length (rownames (df)))])rownames (df.bk) <- NULL colnames (df.bk) <- "CellNumber" $ type <- "Whole dataset Genes" <- rbind (df,df.bk)<- ddply (df, "type" , summarise, grp.mean= mean (CellNumber))ggplot (df,aes (x= CellNumber,fill= type))+ scale_fill_manual (values= c ("#E69F00" , "#56B4E9" ))+ geom_histogram (aes (y= ..density..), position= "identity" , alpha= 0.5 ,bins = 25 )+ xlim (0 ,4800 )+ geom_vline (data= mu, aes (xintercept= grp.mean, color= type),linetype= "dashed" )+ geom_density (alpha= 0.6 )+ scale_y_continuous (labels = percent, name = "percent of genes" ) + theme_classic ()+ theme (legend.position= "bottom" )
set.seed (111 )<- sample (genes.to.test,size = 12 )
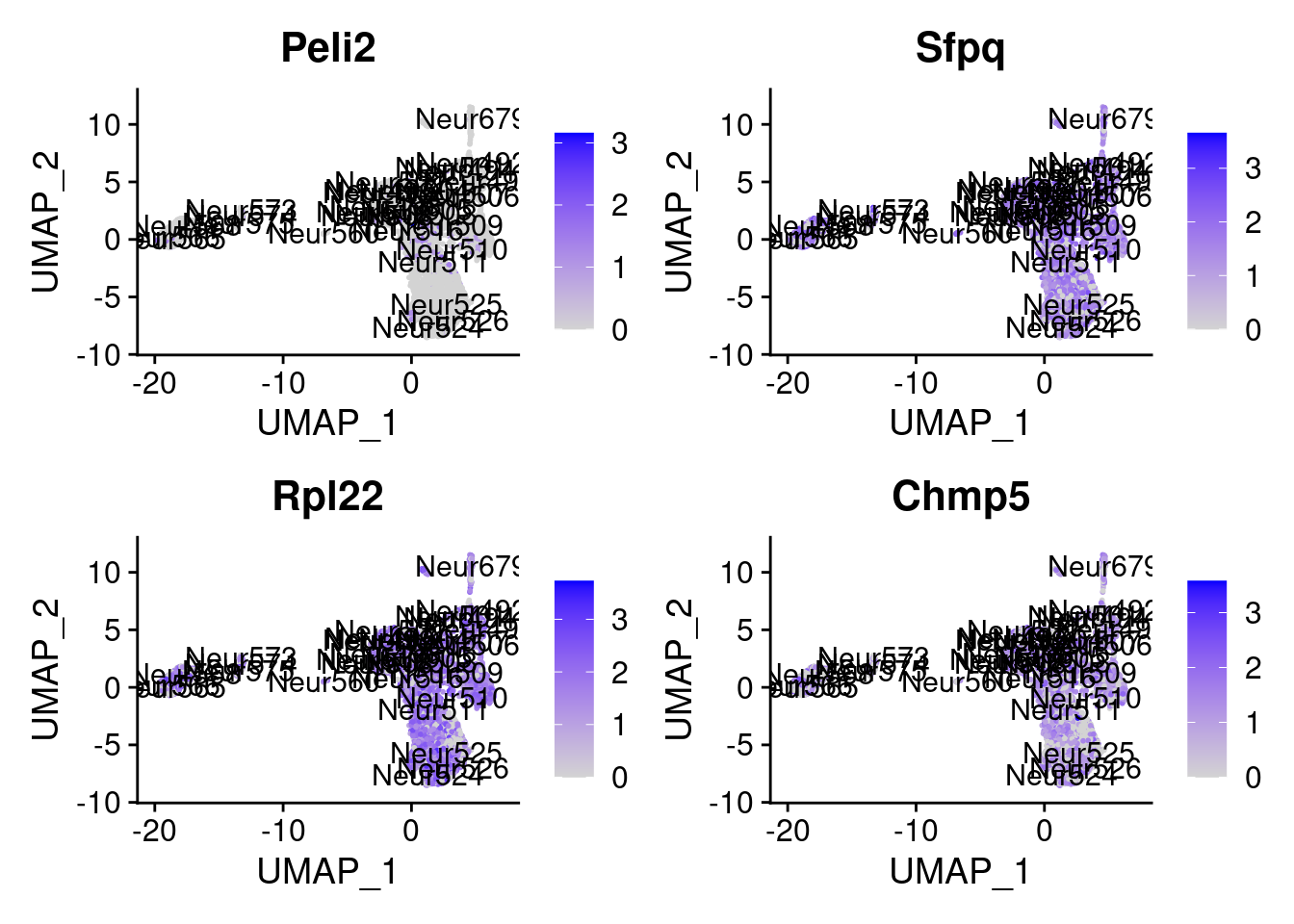
[1] "Grpel1" "Ssna1" "X1110004E09Rik" "Mri1"
[5] "Sod2" "Bola1" "Ppp4c" "Draxin"
[9] "Peli2" "Sfpq" "Rpl22" "Chmp5"
= 0 for (g in c (0 : 2 )) {= g* 4 plot (FeaturePlot (seurat.obj,features = genes[n+ c (1 : 4 )], label = T))
Generally if we look at the genes specifically detected by Seurat, they don’t seems so distinctive for CR cells.
sum (genes.to.test %in% markers.listNeur509$ ` Cortical or hippocampal glutamatergic Seurat ` )/ length (genes.to.test)
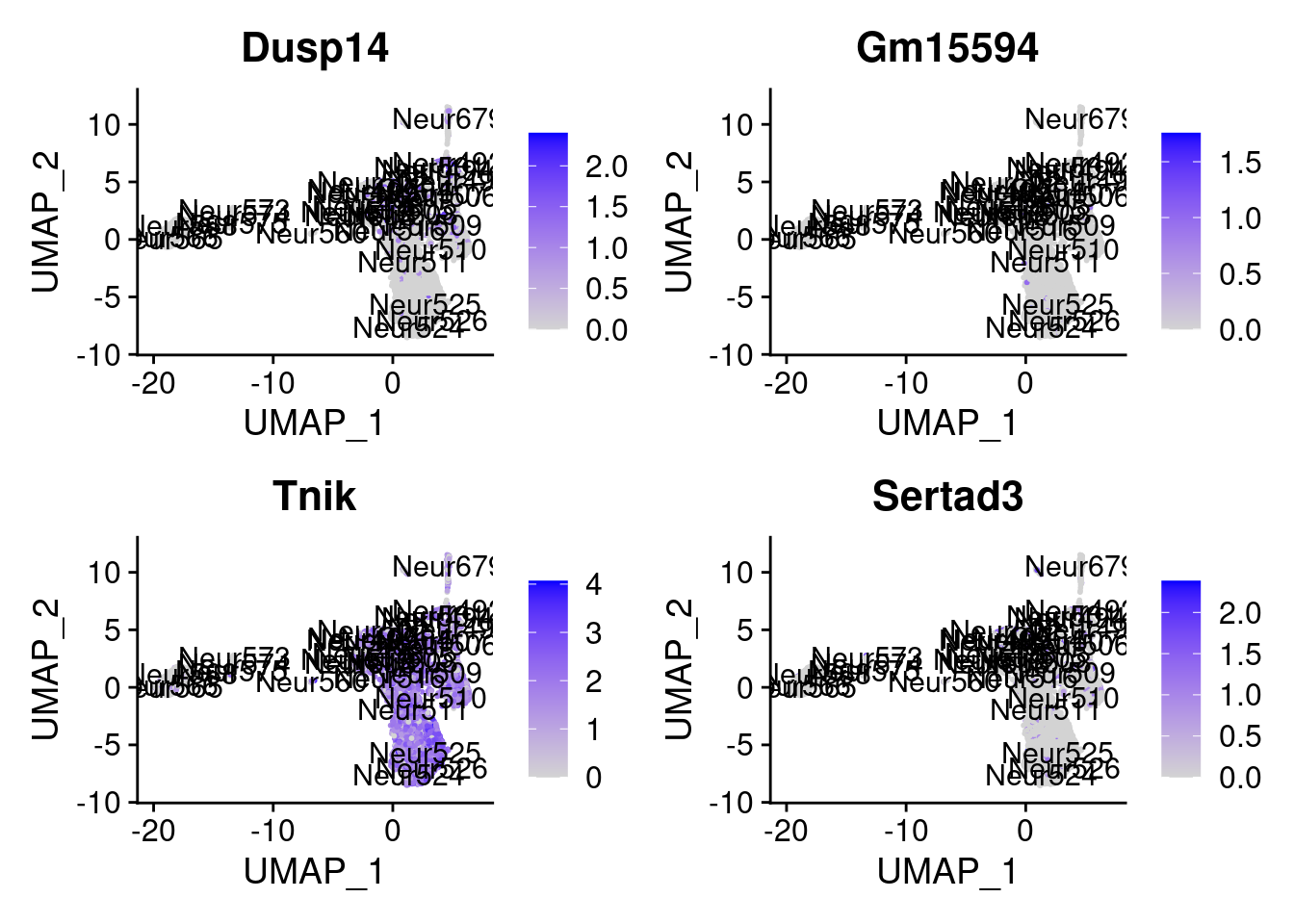
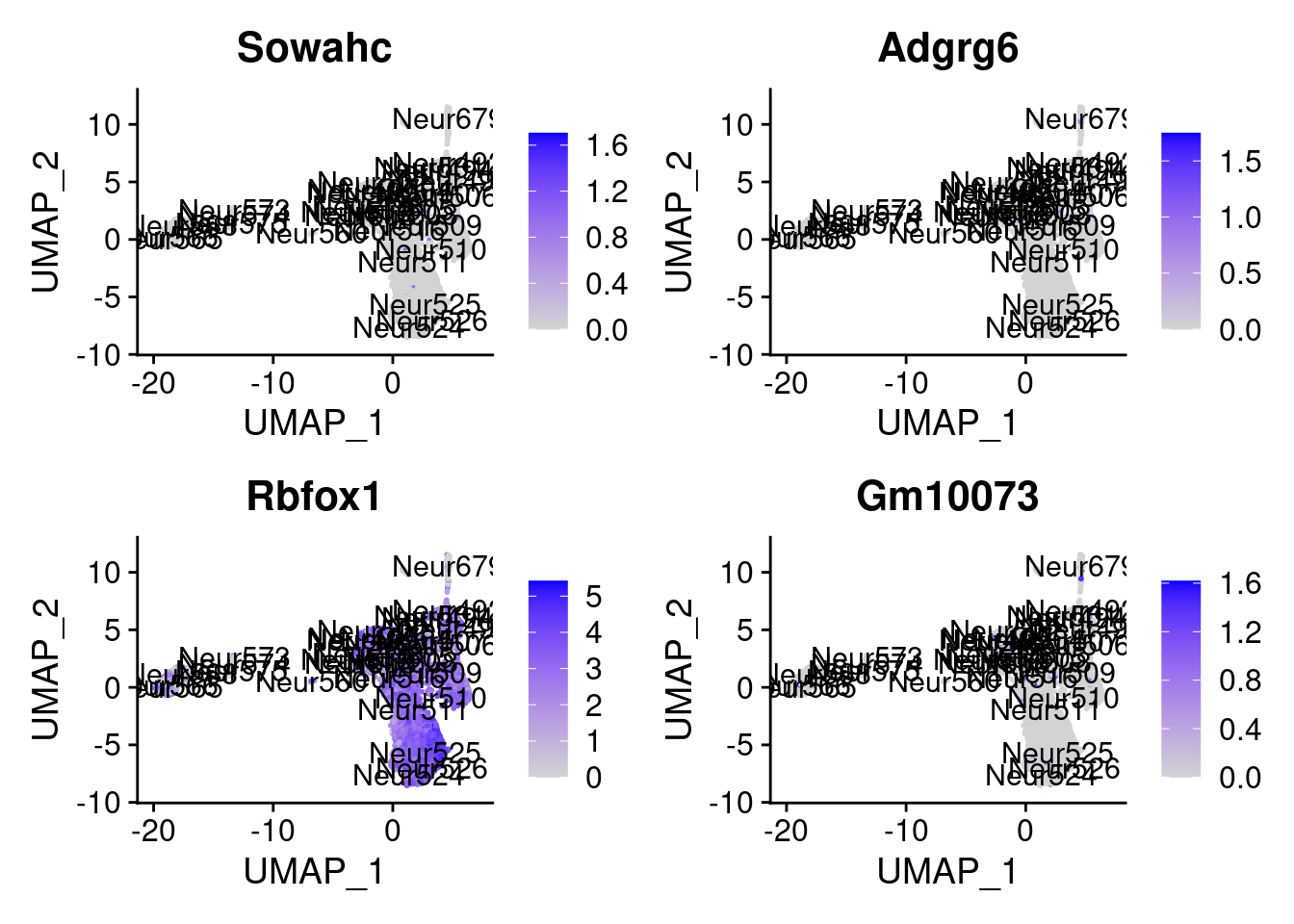
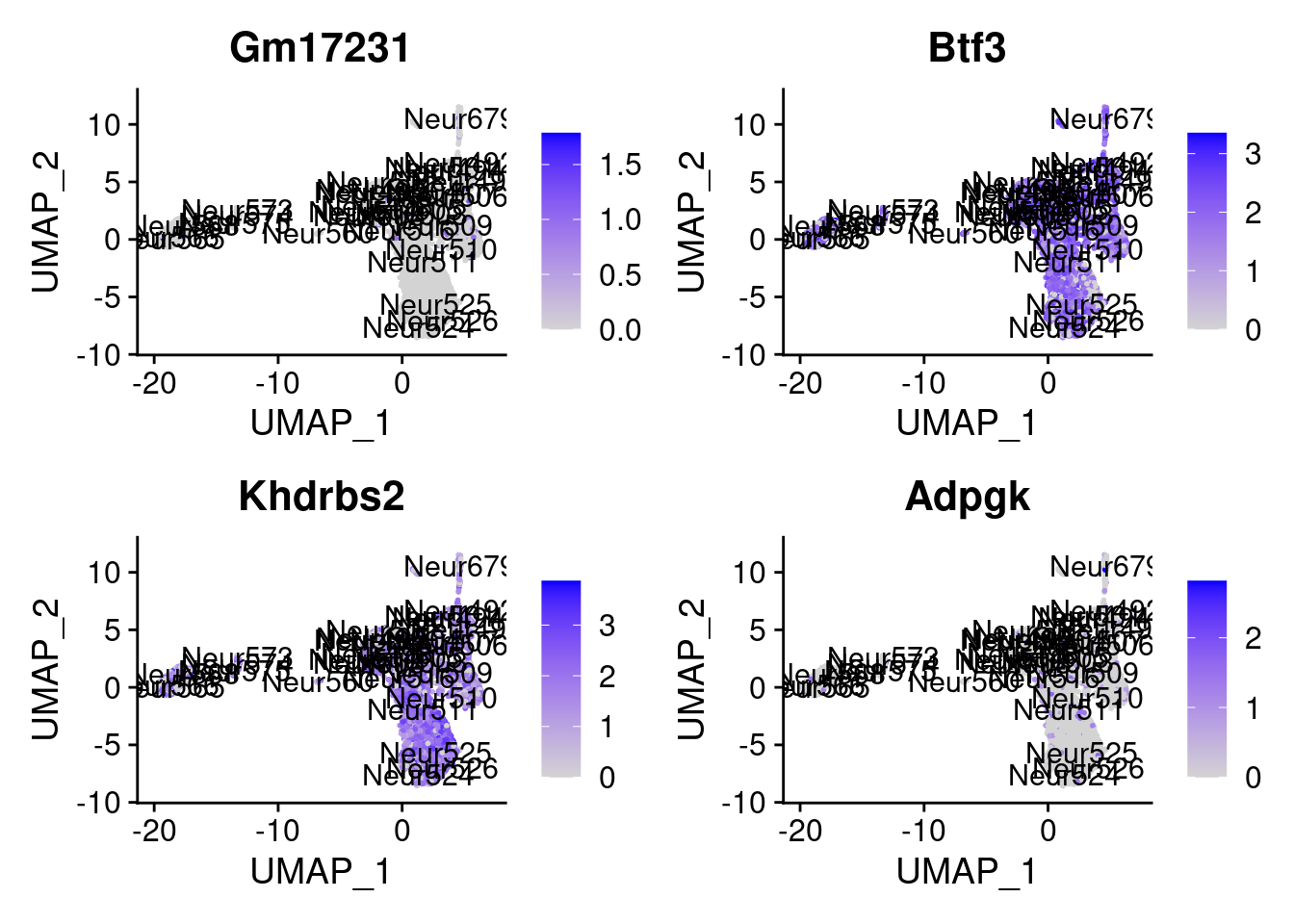
<- markers.listNeur509$ ` Neur509 Cotan ` [! markers.listNeur509$ ` Neur509 Cotan ` %in% markers.listNeur509$ ` Neur509 Seurat ` ]set.seed (11 )<- sample (genes.to.test.Cotan,size = 30 )
[1] "Dusp14" "Gm15594" "Tnik" "Sertad3" "Sowahc"
[6] "Adgrg6" "Rbfox1" "Gm10073" "Gm17231" "Btf3"
[11] "Khdrbs2" "Adpgk" "Pih1d2" "Capzb" "Cox6b1"
[16] "Chst5" "Dcxr" "Tfdp2" "Plcxd2" "Prss41"
[21] "Kcnq5" "AC152827.1" "Atg4a" "Fezf2" "Gm15489"
[26] "Cth" "Zbtb44" "Sec61a1" "Rbm12b1" "Hk2"
= 0 for (g in c (0 : 2 )) {= g* 4 plot (FeaturePlot (seurat.obj,features = genes[n+ c (1 : 4 )], label = T))
<- getNumOfExpressingCells (fb150Obj)[genes.to.test.Cotan]<- as.data.frame (df)colnames (df) <- "CellNumber" rownames (df) <- NULL $ type <- "Only Cotan Genes" <- as.data.frame (getNumOfExpressingCells (fb150Obj)[sample (getGenes (fb150Obj), size = length (rownames (df)))])rownames (df.bk) <- NULL colnames (df.bk) <- "CellNumber" $ type <- "Whole dataset Genes" <- rbind (df,df.bk)<- ddply (df, "type" , summarise, grp.mean= mean (CellNumber))ggplot (df,aes (x= CellNumber,fill= type))+ scale_fill_manual (values= c ("#C69AFF" , "#56B4E9" ))+ geom_histogram (aes (y= ..density..), position= "identity" , alpha= 0.5 ,bins = 25 )+ xlim (0 ,4800 )+ geom_vline (data= mu, aes (xintercept= grp.mean, color= type),linetype= "dashed" )+ geom_density (alpha= 0.6 )+ scale_y_continuous (labels = percent, name = "percent" ) + theme_classic ()+ theme (legend.position= "bottom" )
Now we can test the enrichment for the specific gene both in Seurat and in Cotan.
To make a comparison more equal we select the same number of genes (depending on the smallest group)
<- seurat.obj.markers.ClusterName[seurat.obj.markers.ClusterName$ cluster == "Neur509" & seurat.obj.markers.ClusterName$ gene %in% genes.to.test,]$ gene[1 : length (genes.to.test.Cotan)]<- enrichr (genes.to.testTop, dbs)
Uploading data to Enrichr... Done.
Querying ARCHS4_Tissues... Done.
Parsing results... Done.
plotEnrich (enriched[[1 ]], showTerms = 10 , numChar = 40 , y = "Count" , orderBy = "P.value" )
set.seed (123 )<- data.frame (Terms = str_split (enriched[[1 ]]$ Term[1 : 20 ],pattern = " CL" ,simplify = T )[,1 ],Scores = enriched[[1 ]]$ Combined.Score[1 : 20 ])wordcloud (wordcloud_data$ Terms, wordcloud_data$ Scores, scale = c (3 , 1 ), min.freq = 1 , random.order= FALSE , rot.per= 0.1 ,colors= brewer.pal (8 , "Dark2" ))
<- enrichr (genes.to.test.Cotan, dbs)
Uploading data to Enrichr... Done.
Querying ARCHS4_Tissues... Done.
Parsing results... Done.
plotEnrich (enriched[[1 ]], showTerms = 10 , numChar = 40 , y = "Count" , orderBy = "P.value" )
set.seed (123 )<- data.frame (Terms = str_split (enriched[[1 ]]$ Term[1 : 20 ],pattern = " CL" ,simplify = T )[,1 ],Scores = enriched[[1 ]]$ Combined.Score[1 : 20 ])wordcloud (wordcloud_data$ Terms, wordcloud_data$ Scores, scale = c (3 , 1 ), min.freq = 1 , random.order= FALSE , rot.per= 0.1 ,colors= brewer.pal (8 , "Dark2" ))
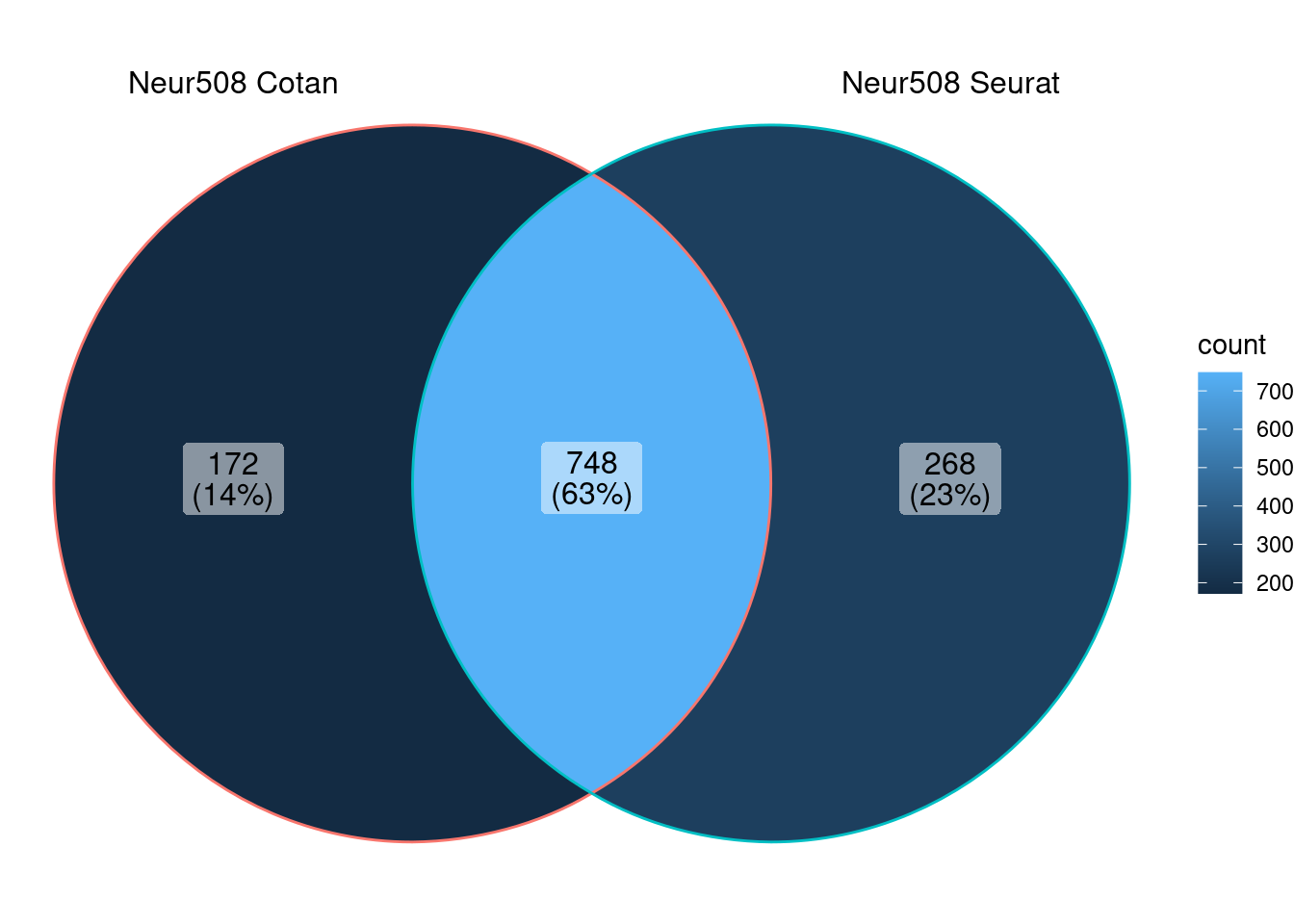
Cluster Neur508
# markers.list.namesNeur508 <- col_concat(crossing(colnames(dea.ClusterName$coex),c("Seurat","Cotan")),sep = " ") # # markers.listNeur508 <- vector("list", length(markers.list.namesNeur508)) # names(markers.listNeur508) <- markers.list.namesNeur508 <- list ("Neur508 Cotan" = NA ,"Neur508 Seurat" = NA )# I take positive coex significant genes for (cl.name in "Neur508" ) {<- rownames (dea.ClusterName$ coex[dea.ClusterName$ ` p-value ` [,cl.name] < 0.01 & dea.ClusterName$ coex[,cl.name] > 0 ,])paste0 (cl.name," Cotan" )]] <- genes#For seurat for (cl.name in "Neur508" ) {<- seurat.obj.markers.ClusterName[seurat.obj.markers.ClusterName$ cluster == cl.name & seurat.obj.markers.ClusterName$ p_val < 0.01 ,]$ genepaste0 (cl.name," Seurat" )]] <- genes
ggVennDiagram (markers.listNeur508)
We can observe that there is a good overlap among the detected markers.
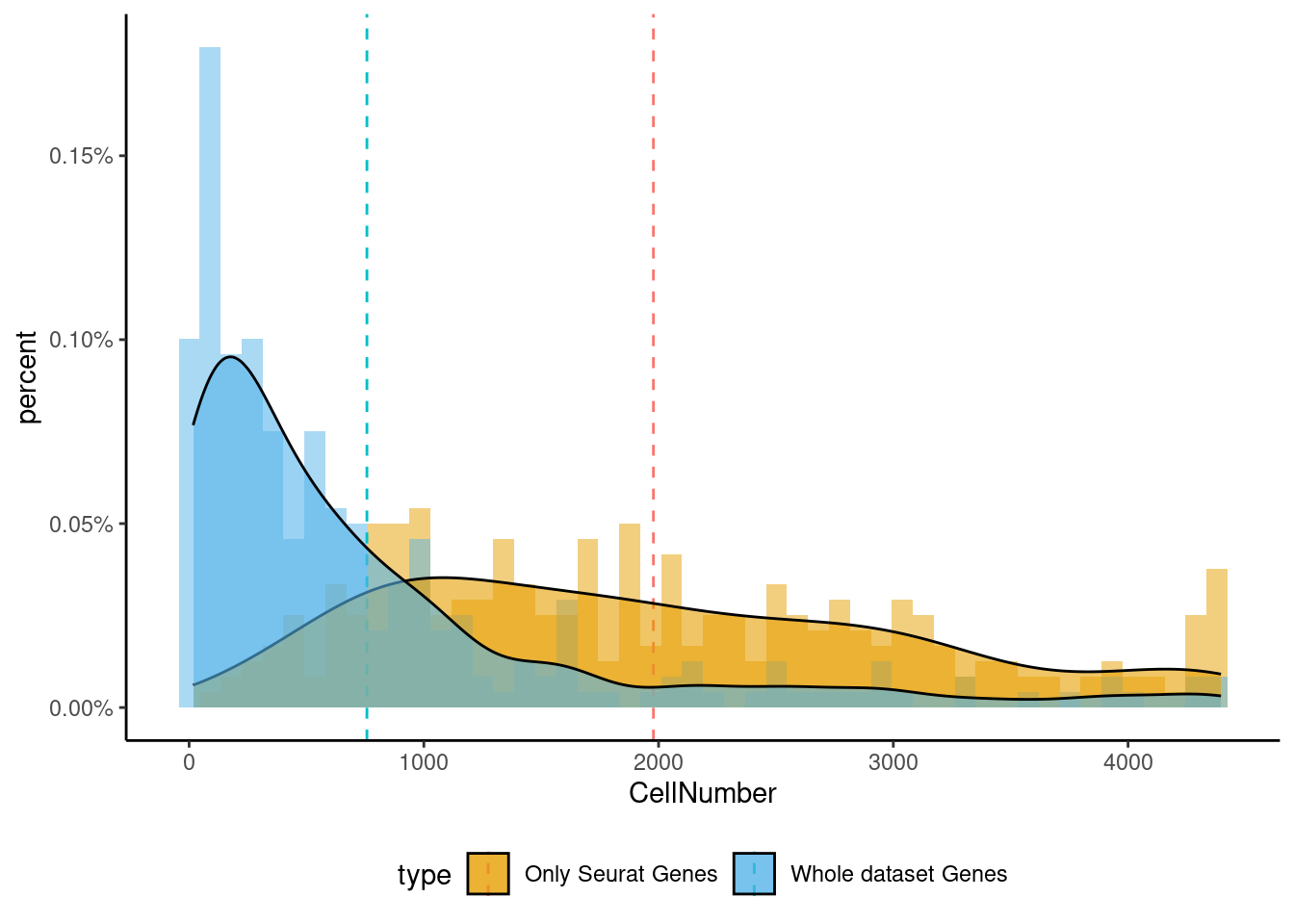
<- markers.listNeur508$ ` Neur508 Seurat ` [! markers.listNeur508$ ` Neur508 Seurat ` %in% markers.listNeur508$ ` Neur508 Cotan ` ]<- getNumOfExpressingCells (fb150Obj)[genes.to.test]<- as.data.frame (df)colnames (df) <- "CellNumber" rownames (df) <- NULL $ type <- "Only Seurat Genes" <- as.data.frame (getNumOfExpressingCells (fb150Obj)[sample (getGenes (fb150Obj), size = length (rownames (df)))])rownames (df.bk) <- NULL colnames (df.bk) <- "CellNumber" $ type <- "Whole dataset Genes" <- rbind (df,df.bk)<- ddply (df, "type" , summarise, grp.mean= mean (CellNumber))ggplot (df,aes (x= CellNumber,fill= type))+ scale_fill_manual (values= c ("#E69F00" , "#56B4E9" ))+ geom_histogram (aes (y= ..density..), position= "identity" , alpha= 0.5 ,bins = 50 )+ geom_vline (data= mu, aes (xintercept= grp.mean, color= type),linetype= "dashed" )+ geom_density (alpha= 0.6 )+ scale_y_continuous (labels = percent, name = "percent" ) + theme_classic ()+ theme (legend.position= "bottom" )
set.seed (111 )<- sample (genes.to.test,size = 12 )
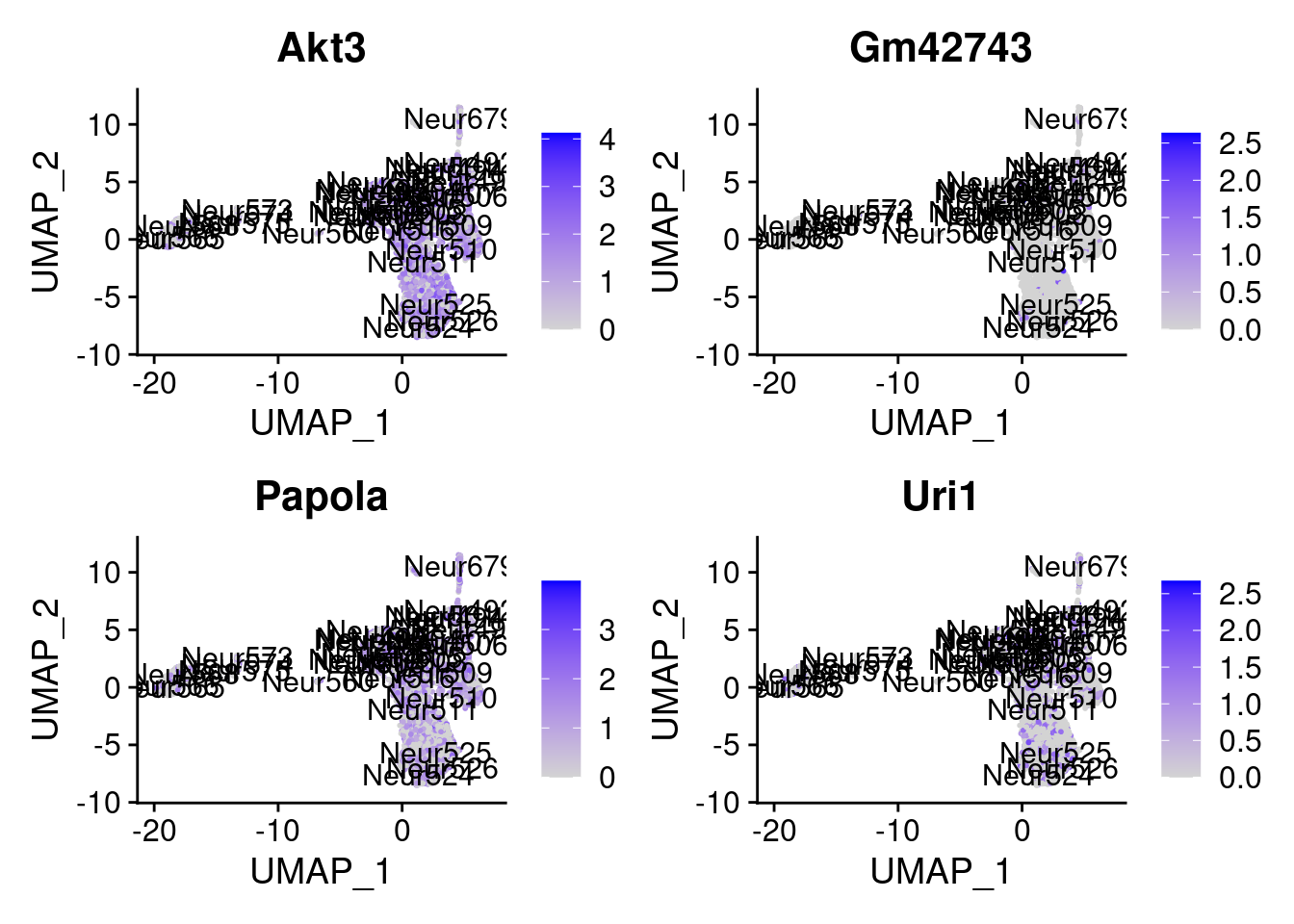
[1] "Gm10131" "Topors" "Agfg1" "Pfdn4" "Arhgdia" "Fam96b" "Kif5c"
[8] "Rpl18" "Akt3" "Gm42743" "Papola" "Uri1"
= 0 for (g in c (0 : 2 )) {= g* 4 plot (FeaturePlot (seurat.obj,features = genes[n+ c (1 : 4 )], label = T))
Generally if we look at the genes specifically detected by Seurat, they don’t seems so distinctive for CR cells.
sum (genes.to.test %in% markers.listNeur508$ ` Cortical or hippocampal glutamatergic Seurat ` )/ length (genes.to.test)
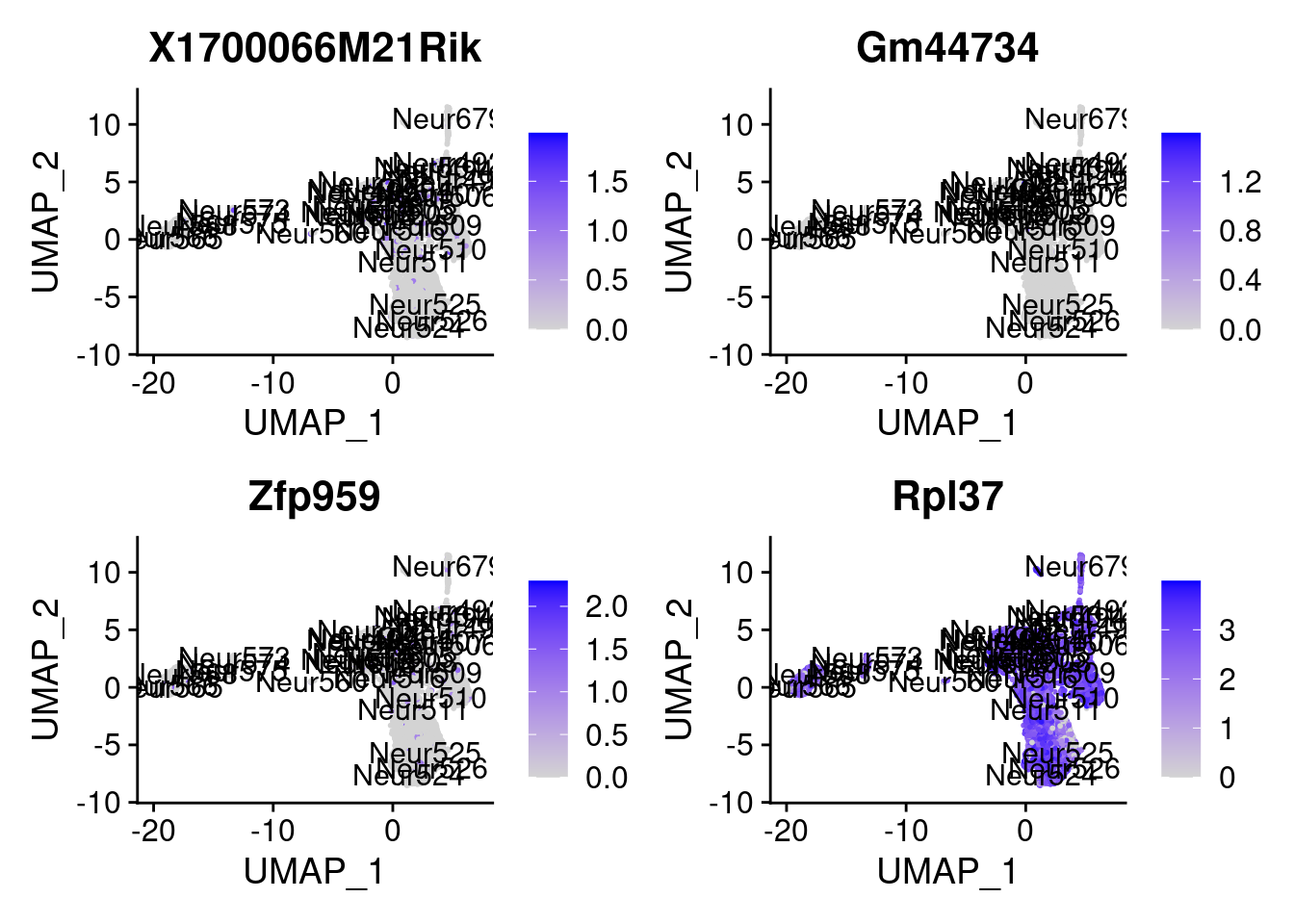
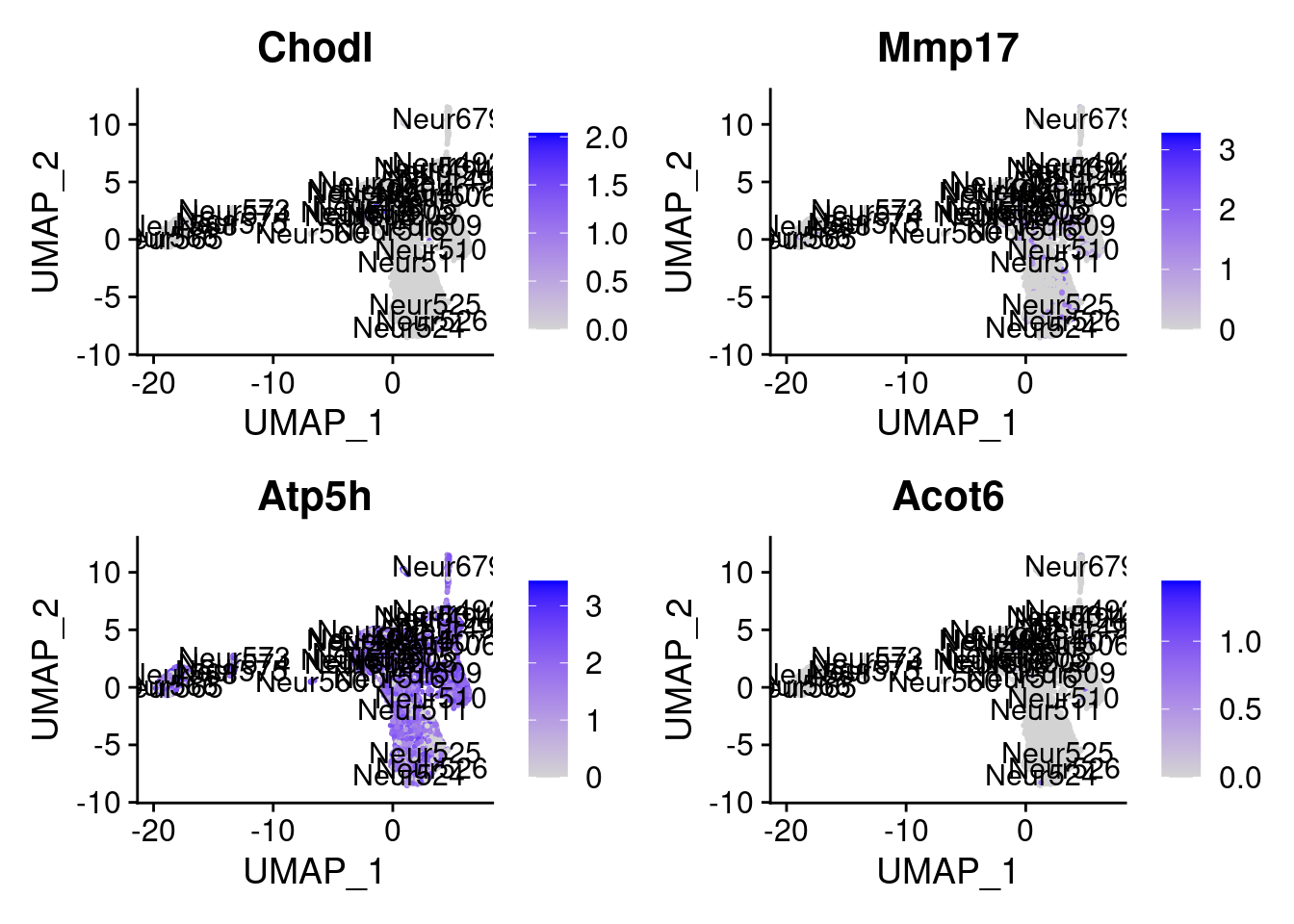
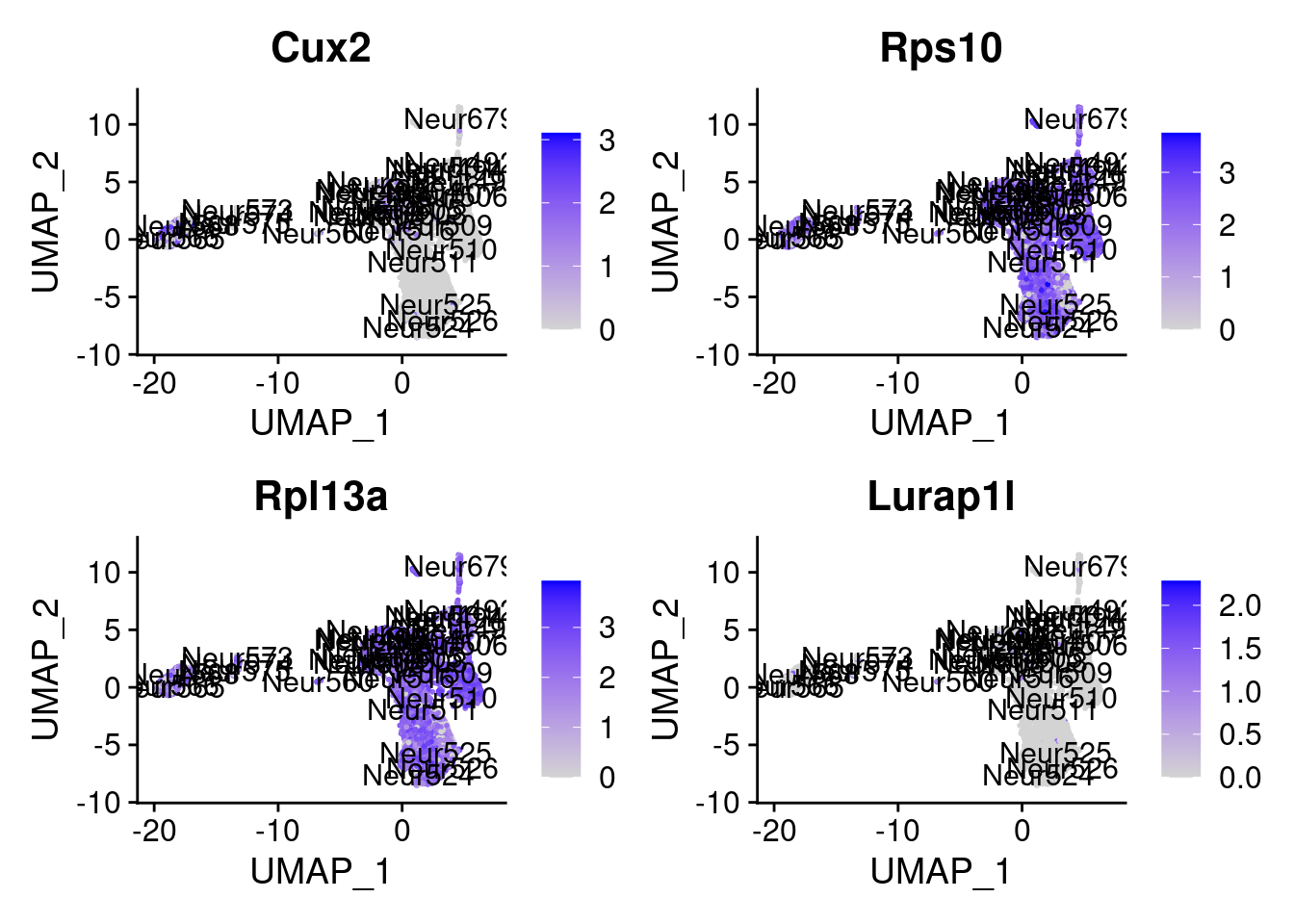
<- markers.listNeur508$ ` Neur508 Cotan ` [! markers.listNeur508$ ` Neur508 Cotan ` %in% markers.listNeur508$ ` Neur508 Seurat ` ]set.seed (11 )<- sample (genes.to.test.Cotan,size = 30 )
[1] "X1700066M21Rik" "Gm44734" "Zfp959" "Rpl37"
[5] "Chodl" "Mmp17" "Atp5h" "Acot6"
[9] "Cux2" "Rps10" "Rpl13a" "Lurap1l"
[13] "Tcf12" "Sirt6" "Gm12184" "Etohd2"
[17] "Laptm4b" "Pomc" "Rpl9" "Inhbb"
[21] "Gpr62" "Sstr3" "D830035M03Rik" "Eef1b2"
[25] "Bmpr1b" "Rps4x" "Tpd52" "Selenow"
[29] "Pla2g7" "Kcnj11"
= 0 for (g in c (0 : 2 )) {= g* 4 plot (FeaturePlot (seurat.obj,features = genes[n+ c (1 : 4 )], label = T))
<- getNumOfExpressingCells (fb150Obj)[genes.to.test.Cotan]<- as.data.frame (df)colnames (df) <- "CellNumber" rownames (df) <- NULL $ type <- "Only Cotan Genes" <- as.data.frame (getNumOfExpressingCells (fb150Obj)[sample (getGenes (fb150Obj), size = length (rownames (df)))])rownames (df.bk) <- NULL colnames (df.bk) <- "CellNumber" $ type <- "Whole dataset Genes" <- rbind (df,df.bk)<- ddply (df, "type" , summarise, grp.mean= mean (CellNumber))ggplot (df,aes (x= CellNumber,fill= type))+ scale_fill_manual (values= c ("#C69AFF" , "#56B4E9" ))+ geom_histogram (aes (y= ..density..), position= "identity" , alpha= 0.5 ,bins = 25 )+ xlim (0 ,4800 )+ geom_vline (data= mu, aes (xintercept= grp.mean, color= type),linetype= "dashed" )+ geom_density (alpha= 0.6 )+ scale_y_continuous (labels = percent, name = "percent" ) + theme_classic ()+ theme (legend.position= "bottom" )
Now we can test the enrichment for the specific gene both in Seurat and in Cotan.
To make a comparison more equal we select the same number of genes (depending on the smallest group)
#genes.to.testTop <- seurat.obj.markers.ClusterName[seurat.obj.markers.ClusterName$cluster == "Neur508" & seurat.obj.markers.ClusterName$gene %in% genes.to.test,]$gene[1:length(genes.to.test.Cotan)] <- enrichr (genes.to.test, dbs)
Uploading data to Enrichr... Done.
Querying ARCHS4_Tissues... Done.
Parsing results... Done.
plotEnrich (enriched[[1 ]], showTerms = 10 , numChar = 40 , y = "Count" , orderBy = "P.value" )
set.seed (123 )<- data.frame (Terms = str_split (enriched[[1 ]]$ Term[1 : 20 ],pattern = " CL" ,simplify = T )[,1 ],Scores = enriched[[1 ]]$ Combined.Score[1 : 20 ])wordcloud (wordcloud_data$ Terms, wordcloud_data$ Scores, scale = c (3 , 1 ), min.freq = 1 , random.order= FALSE , rot.per= 0.1 ,colors= brewer.pal (8 , "Dark2" ))
<- dea.ClusterName$ ` p-value ` [rownames (dea.ClusterName$ ` p-value ` ) %in% genes.to.test.Cotan,]<- rownames (subset.pval[order (subset.pval$ Neur492,decreasing = F),])[1 : length (genes.to.test)]<- enrichr (genes.to.test.Cotan.Top, dbs)
Uploading data to Enrichr... Done.
Querying ARCHS4_Tissues... Done.
Parsing results... Done.
plotEnrich (enriched[[1 ]], showTerms = 10 , numChar = 40 , y = "Count" , orderBy = "P.value" )
set.seed (123 )<- data.frame (Terms = str_split (enriched[[1 ]]$ Term[1 : 20 ],pattern = " CL" ,simplify = T )[,1 ],Scores = enriched[[1 ]]$ Combined.Score[1 : 20 ])wordcloud (wordcloud_data$ Terms, wordcloud_data$ Scores, scale = c (3 , 1 ), min.freq = 1 , random.order= FALSE , rot.per= 0.1 ,colors= brewer.pal (8 , "Dark2" ))
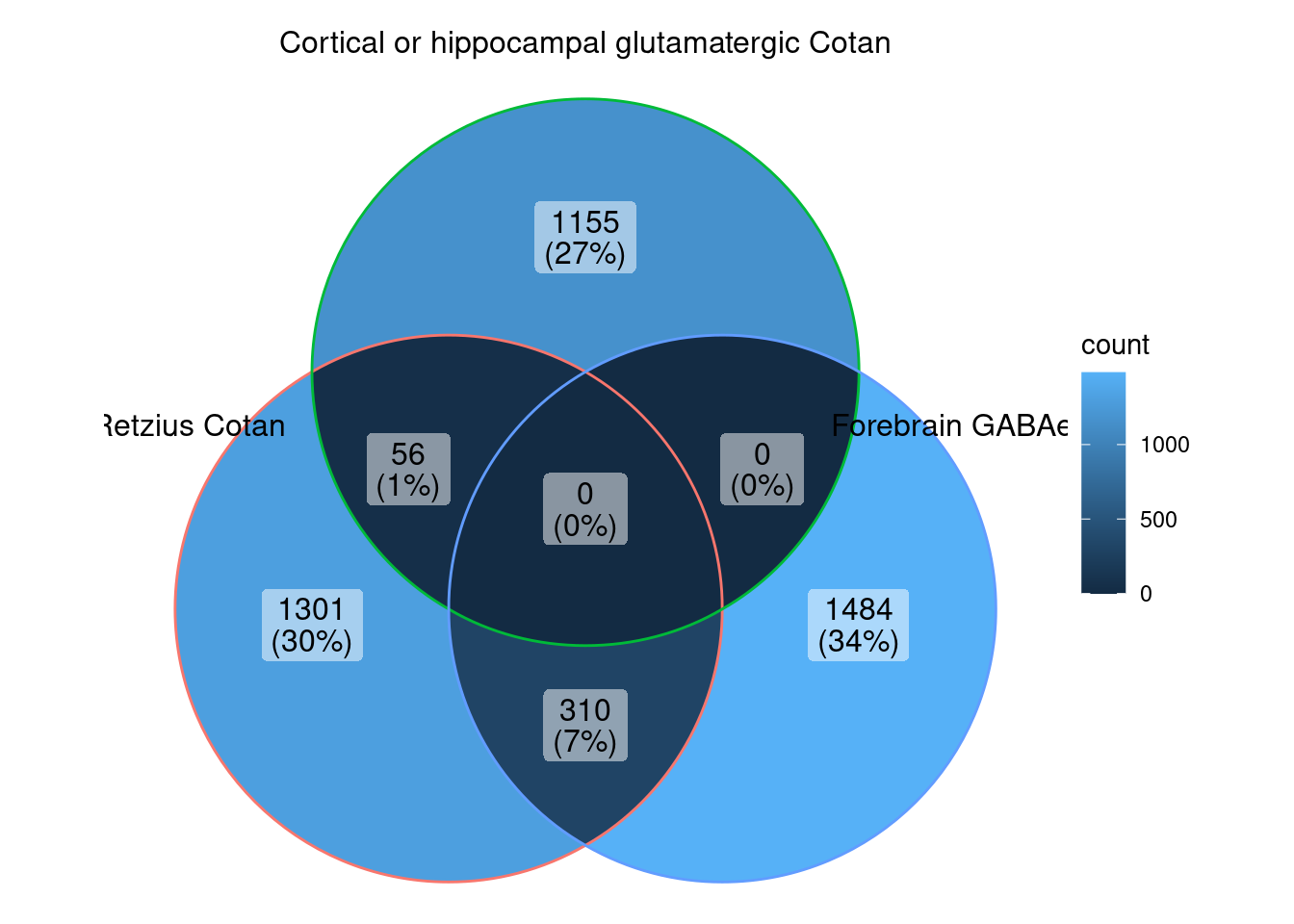
ggVennDiagram (markers.list[c (1 ,3 ,5 )])
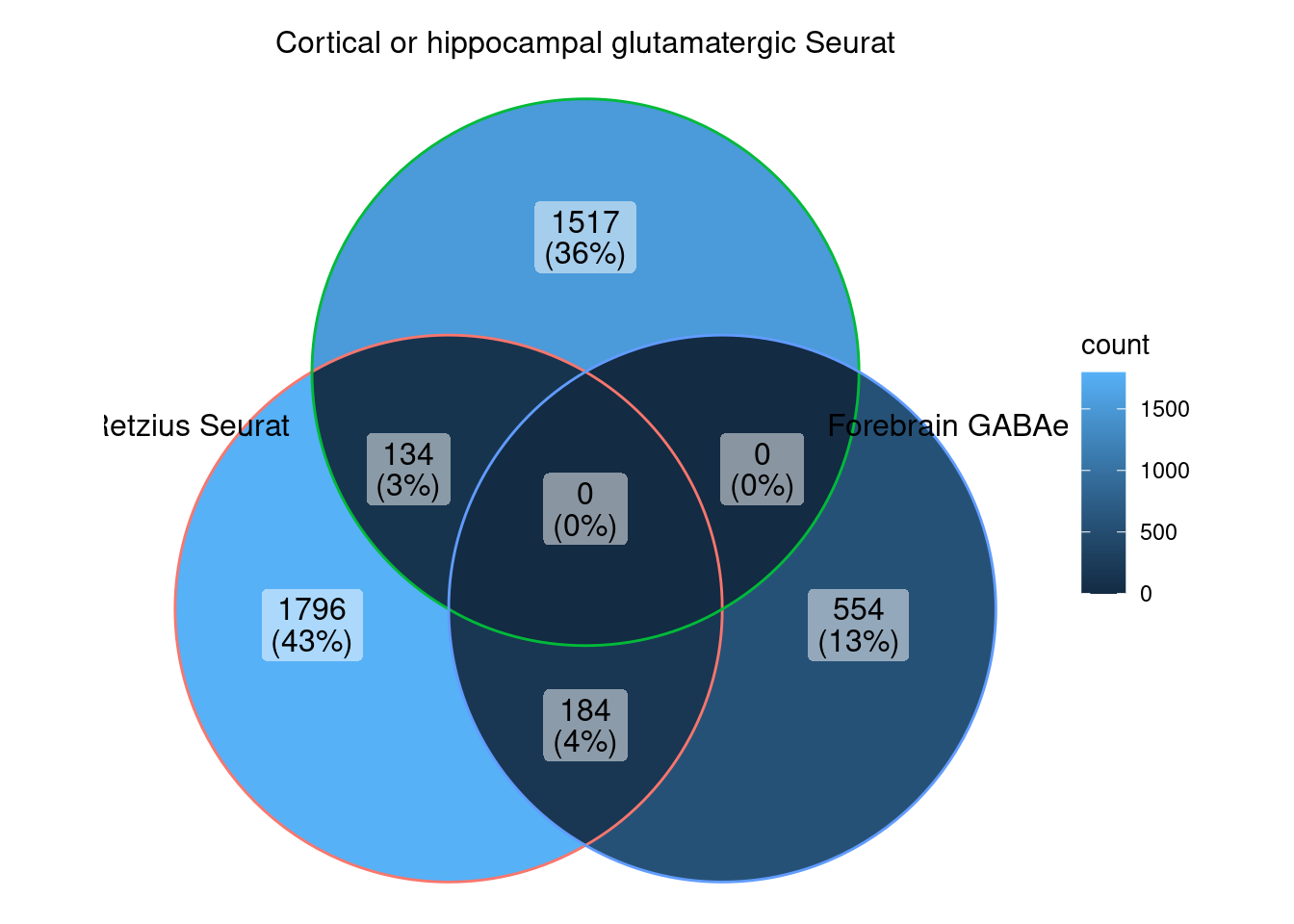
ggVennDiagram (markers.list[c (2 ,4 ,6 )])
R version 4.3.0 (2023-04-21)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.6 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.9.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.9.0
locale:
[1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
[4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
[7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
time zone: Europe/Berlin
tzcode source: system (glibc)
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] enrichR_3.2 tidyr_1.3.0 ggVennDiagram_1.2.2
[4] assertr_3.0.0 stringr_1.5.0 wordcloud_2.6
[7] RColorBrewer_1.1-3 SeuratObject_4.1.3 Seurat_4.3.0
[10] rlang_1.1.0 scales_1.2.1 plyr_1.8.8
[13] COTAN_2.1.5 zeallot_0.1.0 tibble_3.2.1
[16] ggplot2_3.4.2
loaded via a namespace (and not attached):
[1] RcppAnnoy_0.0.20 splines_4.3.0 later_1.3.0
[4] polyclip_1.10-4 factoextra_1.0.7 lifecycle_1.0.3
[7] sf_1.0-13 doParallel_1.0.17 globals_0.16.2
[10] lattice_0.21-8 MASS_7.3-59 dendextend_1.17.1
[13] magrittr_2.0.3 limma_3.56.0 plotly_4.10.1
[16] rmarkdown_2.21 yaml_2.3.7 httpuv_1.6.9
[19] sctransform_0.3.5 askpass_1.1 sp_1.6-0
[22] spatstat.sparse_3.0-1 reticulate_1.30 cowplot_1.1.1
[25] pbapply_1.7-0 DBI_1.1.3 abind_1.4-5
[28] Rtsne_0.16 purrr_1.0.1 BiocGenerics_0.46.0
[31] WriteXLS_6.4.0 circlize_0.4.15 IRanges_2.34.0
[34] S4Vectors_0.38.0 ggrepel_0.9.3 irlba_2.3.5.1
[37] listenv_0.9.0 spatstat.utils_3.0-3 units_0.8-2
[40] umap_0.2.10.0 goftest_1.2-3 RSpectra_0.16-1
[43] spatstat.random_3.1-4 fitdistrplus_1.1-8 parallelly_1.36.0
[46] leiden_0.4.3 codetools_0.2-19 tidyselect_1.2.0
[49] shape_1.4.6 farver_2.1.1 viridis_0.6.2
[52] matrixStats_1.0.0 stats4_4.3.0 spatstat.explore_3.2-1
[55] jsonlite_1.8.4 GetoptLong_1.0.5 e1071_1.7-13
[58] ellipsis_0.3.2 progressr_0.13.0 ggridges_0.5.4
[61] survival_3.5-5 iterators_1.0.14 foreach_1.5.2
[64] tools_4.3.0 ica_1.0-3 Rcpp_1.0.10
[67] glue_1.6.2 gridExtra_2.3 xfun_0.39
[70] ggthemes_4.2.4 dplyr_1.1.2 withr_2.5.0
[73] fastmap_1.1.1 fansi_1.0.4 openssl_2.0.6
[76] digest_0.6.31 parallelDist_0.2.6 R6_2.5.1
[79] mime_0.12 colorspace_2.1-0 scattermore_1.2
[82] tensor_1.5 spatstat.data_3.0-1 utf8_1.2.3
[85] generics_0.1.3 data.table_1.14.8 class_7.3-21
[88] httr_1.4.5 htmlwidgets_1.6.2 uwot_0.1.14
[91] pkgconfig_2.0.3 gtable_0.3.3 ComplexHeatmap_2.16.0
[94] lmtest_0.9-40 htmltools_0.5.5 clue_0.3-64
[97] png_0.1-8 knitr_1.42 rstudioapi_0.14
[100] reshape2_1.4.4 rjson_0.2.21 nlme_3.1-162
[103] curl_5.0.0 proxy_0.4-27 zoo_1.8-12
[106] GlobalOptions_0.1.2 RVenn_1.1.0 KernSmooth_2.23-20
[109] parallel_4.3.0 miniUI_0.1.1.1 vipor_0.4.5
[112] RcppZiggurat_0.1.6 ggrastr_1.0.2 pillar_1.9.0
[115] grid_4.3.0 vctrs_0.6.1 RANN_2.6.1
[118] promises_1.2.0.1 xtable_1.8-4 cluster_2.1.4
[121] beeswarm_0.4.0 evaluate_0.20 cli_3.6.1
[124] compiler_4.3.0 crayon_1.5.2 future.apply_1.11.0
[127] labeling_0.4.2 classInt_0.4-9 ggbeeswarm_0.7.2
[130] stringi_1.7.12 viridisLite_0.4.1 deldir_1.0-6
[133] assertthat_0.2.1 munsell_0.5.0 lazyeval_0.2.2
[136] spatstat.geom_3.2-1 Matrix_1.5-4.1 patchwork_1.1.2
[139] future_1.32.0 shiny_1.7.4 ROCR_1.0-11
[142] Rfast_2.0.7 igraph_1.4.2 RcppParallel_5.1.7