library(COTAN)
library(pROC)
options(parallelly.fork.enable = TRUE)
#library(Seurat)
#library(monocle3)
#library(reticulate)
library(stringr)
library(dplyr)
library(tidyr)
library(ggplot2)
library(ggpubr)
dirOut <- file.path(".", "Results/FDR/")
if (!dir.exists(dirOut)) {
dir.create(dirOut)
}
dataSetDir <- file.path("Data/MouseCortexFromLoom/FDR/", "MergedClusters_For_FDR")Results - Log Fold Change
Preamble
Dataset composition
datasets_csv <- read.csv(file.path(dataSetDir,"Cells_Usage_DataFrame.csv"),
row.names = 1, header = TRUE, quote = '"'
)
datasets_csv[1:10, ] Group Collection E13.5.432 E13.5.187 E13.5.434
1 2_Clusters_even_near E13.5-434_+_E15.0-428 0 0 318
2 2_Clusters_even_near E15.0-432_+_E13.5-432 536 0 0
3 2_Clusters_even_near E15.0-508_+_E15.0-509 0 0 0
4 2_Clusters_even_medium E13.5-187_+_E13.5-184 0 292 0
5 2_Clusters_even_medium E15.0-434_+_E17.5-516 0 0 0
6 2_Clusters_even_medium E15.0-437_+_E15.0-508 0 0 0
7 2_Clusters_even_far E17.5-516_+_E13.5-187 0 297 0
8 2_Clusters_even_far E15.0-510_+_E13.5-437 0 0 0
9 2_Clusters_even_far E15.0-509_+_E13.5-184 0 0 0
10 2_Clusters_uneven_near E13.5-434_+_E15.0-428 0 0 326
E13.5.184 E13.5.437 E13.5.510 E15.0.432 E15.0.509 E15.0.510 E15.0.508
1 0 0 0 0 0 0 0
2 0 0 0 536 0 0 0
3 0 0 0 0 397 0 397
4 292 0 0 0 0 0 0
5 0 0 0 0 0 0 0
6 0 0 0 0 0 0 258
7 0 0 0 0 0 0 0
8 0 259 0 0 0 259 0
9 292 0 0 0 292 0 0
10 0 0 0 0 0 0 0
E15.0.428 E15.0.434 E15.0.437 E17.5.516 E17.5.505
1 318 0 0 0 0
2 0 0 0 0 0
3 0 0 0 0 0
4 0 0 0 0 0
5 0 273 0 273 0
6 0 0 258 0 0
7 0 0 0 297 0
8 0 0 0 0 0
9 0 0 0 0 0
10 37 0 0 0 0Define which genes are expressed
For each data set we need to define, independently from the DEA methods, which genes are specific for each cluster. So we need to define first which genes are expressed and which are not expressed. To do so we can take advantage from the fact that we have the original clusters from which the cells were sampled to create the artificial datasets. So looking to the original cluster we define as expressed all genes present for which there are at least 2 reads every 10 cells. We define as enriched for a cluster the genes that are present and have at least 3 times as many average reads as the average reads outside the cluster.
readsLogMeansPerCluster <- readRDS(file.path(dirOut, "readsLogAverageCount_PerCluster.RDS"))
thresholdLFC <- log10(3.0) # presence if 3 times more reads on average
thresholdLFM <- log10(10000 * 2 / 10) # 2 reads each 10 cells
10^thresholdLFC[1] 310^(thresholdLFM-4)[1] 0.22 Clusters even
2_Clusters_even_near
True vector
subset.datasets_csv <-datasets_csv[datasets_csv$Group == "2_Clusters_even_near", ]
ground_truth_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
clusters <- str_split(subset.datasets_csv$Collection[ind], pattern = "_[+]_", simplify = T)
reads.LM.subset <- readsLogMeansPerCluster[, clusters]
ground_truth <- as.data.frame(matrix(nrow = nrow(reads.LM.subset),
ncol = ncol(reads.LM.subset)))
rownames(ground_truth) <- rownames(reads.LM.subset)
colnames(ground_truth) <- colnames(reads.LM.subset)
for (col in 1:ncol(ground_truth)) {
# log fold change
ground_truth[, col] <- reads.LM.subset[, col] - rowMeans(reads.LM.subset[, -col, drop = FALSE])
ground_truth[, col] <- ground_truth[, col] > thresholdLFC & reads.LM.subset[, col] > thresholdLFM
}
ground_truth$genes <- rownames(ground_truth)
ground_truth <- pivot_longer(ground_truth,
cols = 1:(ncol(ground_truth)-1),
names_to = "clusters")
ground_truth$data_set <- subset.datasets_csv[ind, 1]
ground_truth$set_number <- ind
ground_truth_tot <- rbind(ground_truth_tot, ground_truth)
}
ground_truth_tot <- ground_truth_tot[2:nrow(ground_truth_tot),]
head(ground_truth_tot)# A tibble: 6 × 5
genes clusters value data_set set_number
<chr> <chr> <lgl> <chr> <int>
1 Neil2 E13.5-434 FALSE 2_Clusters_even_near 1
2 Neil2 E15.0-428 FALSE 2_Clusters_even_near 1
3 Lamc1 E13.5-434 FALSE 2_Clusters_even_near 1
4 Lamc1 E15.0-428 FALSE 2_Clusters_even_near 1
5 Lama1 E13.5-434 FALSE 2_Clusters_even_near 1
6 Lama1 E15.0-428 FALSE 2_Clusters_even_near 1length(unique(ground_truth_tot$genes))[1] 14695sum(ground_truth_tot$value)[1] 80ROC for COTAN
onlyPositive.pVal.Cotan_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",
subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
deaCOTAN <- getClusterizationData(dataset,clName = "mergedClusters")[[2]]
pvalCOTAN <- pValueFromDEA(deaCOTAN,
numCells = getNumCells(dataset),adjustmentMethod = "none")
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.names <- c(cl.names,
str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1])
cl.names <- cl.names[!is.na(cl.names)]
}
colnames(deaCOTAN) <- cl.names
colnames(pvalCOTAN) <- cl.names
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
onlyPositive.pVal.Cotan <- pvalCOTAN
for (cl in cl.names) {
print(cl)
#temp.DEA.CotanSign <- deaCOTAN[rownames(pvalCOTAN[pvalCOTAN[,cl] < 0.05,]) ,]
onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl] <- 1 #onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl]+1
}
onlyPositive.pVal.Cotan$genes <- rownames(onlyPositive.pVal.Cotan)
onlyPositive.pVal.Cotan <- pivot_longer(onlyPositive.pVal.Cotan,
values_to = "p_values",
cols = 1:(ncol(onlyPositive.pVal.Cotan)-1),
names_to = "clusters")
onlyPositive.pVal.Cotan$data_set <- subset.datasets_csv[ind,1]
onlyPositive.pVal.Cotan$set_number <- ind
onlyPositive.pVal.Cotan_tot <- rbind(onlyPositive.pVal.Cotan_tot, onlyPositive.pVal.Cotan)
}[1] "E13.5-434"
[1] "E15.0-428"
[1] "E15.0-432"
[1] "E13.5-432"
[1] "E15.0-509"
[1] "E15.0-508"onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[2:nrow(onlyPositive.pVal.Cotan_tot),]
onlyPositive.pVal.Cotan_tot <- merge.data.frame(onlyPositive.pVal.Cotan_tot,
ground_truth_tot,by = c("genes","clusters","data_set","set_number"),all.x = T,all.y = F)
#onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[order(onlyPositive.pVal.Cotan_tot$p_values,
# decreasing = F),]
# df <- as.data.frame(matrix(nrow = nrow(onlyPositive.pVal.Cotan_tot),ncol = 3))
# colnames(df) <- c("TPR","FPR","Method")
# df$Method <- "COTAN"
#
# Positive <- sum(onlyPositive.pVal.Cotan_tot$value)
# Negative <- sum(!onlyPositive.pVal.Cotan_tot$value)
#
# for (i in 1:nrow(onlyPositive.pVal.Cotan_tot)) {
# df[i,"TPR"] <- sum(onlyPositive.pVal.Cotan_tot[1:i,"value"])/Positive
# df[i,"FPR"] <- (i-sum(onlyPositive.pVal.Cotan_tot[1:i,"value"]))/Negative
# Convert TRUE/FALSE to 1/0
onlyPositive.pVal.Cotan_tot$value <- as.numeric(onlyPositive.pVal.Cotan_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultCOTAN <- roc(onlyPositive.pVal.Cotan_tot$value, 1 - onlyPositive.pVal.Cotan_tot$p_values)
# Plot the ROC curve
#plot(roc_resultCOTAN)ROC for Seurat
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat scTransform
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_ScTransform_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_scTr <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat Bimod
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_Bimod_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_Bimod <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Monocle
deaMonocle_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaMonocle <- read.csv(file.path(dirOut,paste0(file.code,"Monocle_DEA_genes.csv")),row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaMonocle[deaMonocle$cell_group == cl.val,"cell_group"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "cell_group",
replacement = "clusters")
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "gene_id",
replacement = "genes")
deaMonocle$data_set <- subset.datasets_csv[ind,1]
deaMonocle$set_number <- ind
deaMonocle <- as.data.frame(deaMonocle)
deaMonocle_tot <- rbind(deaMonocle_tot, deaMonocle)
}
deaMonocle_tot <- deaMonocle_tot[2:nrow(deaMonocle_tot),]
deaMonocle_tot <- merge.data.frame(deaMonocle_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaMonocle_tot$value <- as.numeric(deaMonocle_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultMonocle <- roc(deaMonocle_tot$value, 1 - deaMonocle_tot$marker_test_p_value)
# Plot the ROC curve
#plot(roc_resultMonocle)ROC from ScamPy
deaScamPy_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaScamPy <- read.csv(file.path(dirOut,paste0(file.code,"ScanPy_DEA_genes.csv")),
row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaScamPy[deaScamPy$clusters == paste0("cl",cl.val),"clusters"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
deaScamPy$data_set <- subset.datasets_csv[ind,1]
deaScamPy$set_number <- ind
deaScamPy_tot <- rbind(deaScamPy_tot, deaScamPy)
}
deaScamPy_tot <- deaScamPy_tot[2:nrow(deaScamPy_tot),]
deaScamPy_tot <- merge.data.frame(deaScamPy_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaScamPy_tot$value <- as.numeric(deaScamPy_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultScamPy <- roc(deaScamPy_tot$value, 1 - deaScamPy_tot$pval)
# Plot the ROC curve
#plot(roc_resultScamPy)Summary ROC for all methods
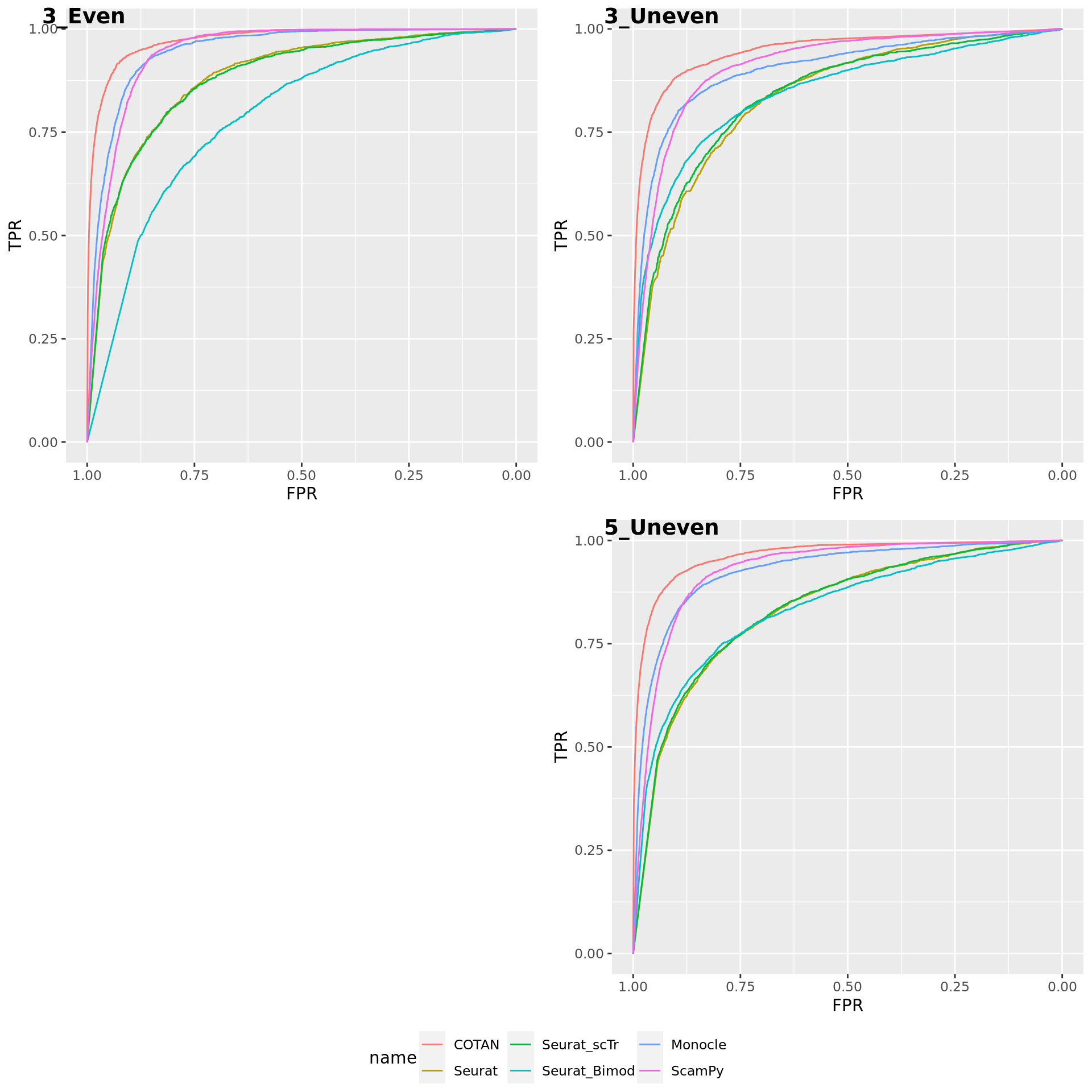
TwoClusters_even_near <- ggroc(list(COTAN=roc_resultCOTAN, Seurat=roc_resultSeurat,
Seurat_scTr = roc_resultSeurat_scTr, Seurat_Bimod = roc_resultSeurat_Bimod,
Monocle=roc_resultMonocle, ScamPy=roc_resultScamPy))
TwoClusters_even_nearPL <- TwoClusters_even_near + xlab("FPR") + ylab("TPR")
TwoClusters_even_nearPL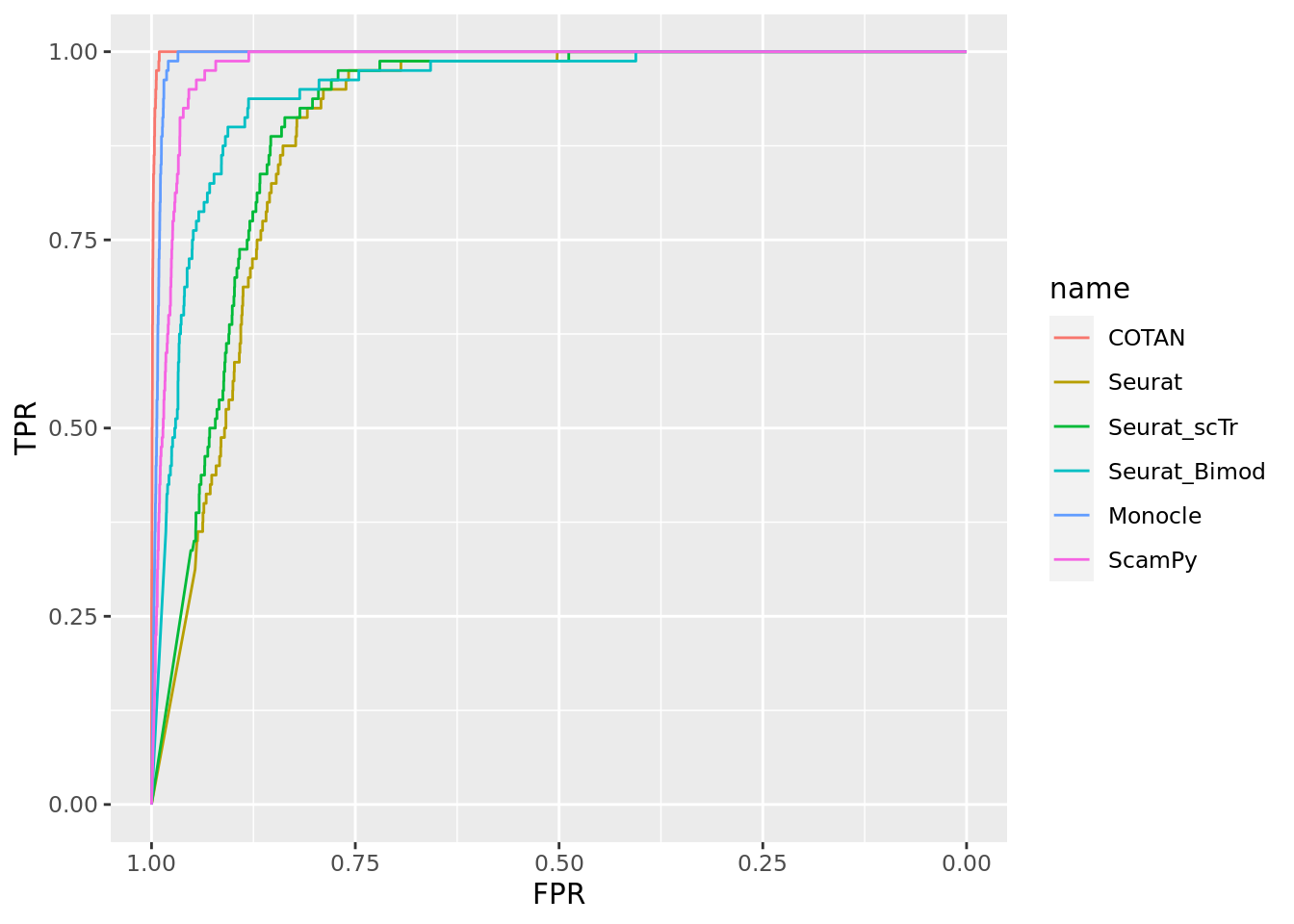
2_Clusters_even_medium
True vector
subset.datasets_csv <-datasets_csv[datasets_csv$Group == "2_Clusters_even_medium", ]
ground_truth_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
clusters <- str_split(subset.datasets_csv$Collection[ind], pattern = "_[+]_", simplify = T)
reads.LM.subset <- readsLogMeansPerCluster[, clusters]
ground_truth <- as.data.frame(matrix(nrow = nrow(reads.LM.subset),
ncol = ncol(reads.LM.subset)))
rownames(ground_truth) <- rownames(reads.LM.subset)
colnames(ground_truth) <- colnames(reads.LM.subset)
for (col in 1:ncol(ground_truth)) {
# log fold change
ground_truth[, col] <- reads.LM.subset[, col] - rowMeans(reads.LM.subset[, -col, drop = FALSE])
ground_truth[, col] <- ground_truth[, col] > thresholdLFC & reads.LM.subset[, col] > thresholdLFM
}
ground_truth$genes <- rownames(ground_truth)
ground_truth <- pivot_longer(ground_truth,
cols = 1:(ncol(ground_truth)-1),
names_to = "clusters")
ground_truth$data_set <- subset.datasets_csv[ind, 1]
ground_truth$set_number <- ind
ground_truth_tot <- rbind(ground_truth_tot, ground_truth)
}
ground_truth_tot <- ground_truth_tot[2:nrow(ground_truth_tot),]
head(ground_truth_tot)# A tibble: 6 × 5
genes clusters value data_set set_number
<chr> <chr> <lgl> <chr> <int>
1 Neil2 E13.5-187 FALSE 2_Clusters_even_medium 1
2 Neil2 E13.5-184 FALSE 2_Clusters_even_medium 1
3 Lamc1 E13.5-187 FALSE 2_Clusters_even_medium 1
4 Lamc1 E13.5-184 FALSE 2_Clusters_even_medium 1
5 Lama1 E13.5-187 FALSE 2_Clusters_even_medium 1
6 Lama1 E13.5-184 FALSE 2_Clusters_even_medium 1length(unique(ground_truth_tot$genes))[1] 14695sum(ground_truth_tot$value)[1] 1559ROC for COTAN
onlyPositive.pVal.Cotan_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",
subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
deaCOTAN <- getClusterizationData(dataset,clName = "mergedClusters")[[2]]
pvalCOTAN <- pValueFromDEA(deaCOTAN,
numCells = getNumCells(dataset),adjustmentMethod = "none")
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.names <- c(cl.names,
str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1])
cl.names <- cl.names[!is.na(cl.names)]
}
colnames(deaCOTAN) <- cl.names
colnames(pvalCOTAN) <- cl.names
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
onlyPositive.pVal.Cotan <- pvalCOTAN
for (cl in cl.names) {
print(cl)
#temp.DEA.CotanSign <- deaCOTAN[rownames(pvalCOTAN[pvalCOTAN[,cl] < 0.05,]) ,]
onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl] <- 1 #onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl]+1
}
onlyPositive.pVal.Cotan$genes <- rownames(onlyPositive.pVal.Cotan)
onlyPositive.pVal.Cotan <- pivot_longer(onlyPositive.pVal.Cotan,
values_to = "p_values",
cols = 1:(ncol(onlyPositive.pVal.Cotan)-1),
names_to = "clusters")
onlyPositive.pVal.Cotan$data_set <- subset.datasets_csv[ind,1]
onlyPositive.pVal.Cotan$set_number <- ind
onlyPositive.pVal.Cotan_tot <- rbind(onlyPositive.pVal.Cotan_tot, onlyPositive.pVal.Cotan)
}[1] "E13.5-187"
[1] "E13.5-184"
[1] "E17.5-516"
[1] "E15.0-434"
[1] "E15.0-508"
[1] "E15.0-437"onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[2:nrow(onlyPositive.pVal.Cotan_tot),]
onlyPositive.pVal.Cotan_tot <- merge.data.frame(onlyPositive.pVal.Cotan_tot,
ground_truth_tot,by = c("genes","clusters","data_set","set_number"),all.x = T,all.y = F)
#onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[order(onlyPositive.pVal.Cotan_tot$p_values,
# decreasing = F),]
# df <- as.data.frame(matrix(nrow = nrow(onlyPositive.pVal.Cotan_tot),ncol = 3))
# colnames(df) <- c("TPR","FPR","Method")
# df$Method <- "COTAN"
#
# Positive <- sum(onlyPositive.pVal.Cotan_tot$value)
# Negative <- sum(!onlyPositive.pVal.Cotan_tot$value)
#
# for (i in 1:nrow(onlyPositive.pVal.Cotan_tot)) {
# df[i,"TPR"] <- sum(onlyPositive.pVal.Cotan_tot[1:i,"value"])/Positive
# df[i,"FPR"] <- (i-sum(onlyPositive.pVal.Cotan_tot[1:i,"value"]))/Negative
# Convert TRUE/FALSE to 1/0
onlyPositive.pVal.Cotan_tot$value <- as.numeric(onlyPositive.pVal.Cotan_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultCOTAN <- roc(onlyPositive.pVal.Cotan_tot$value, 1 - onlyPositive.pVal.Cotan_tot$p_values)
# Plot the ROC curve
#plot(roc_resultCOTAN)ROC for Seurat
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat scTransform
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_ScTransform_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_scTr <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat Bimod
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_Bimod_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_Bimod <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Monocle
deaMonocle_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaMonocle <- read.csv(file.path(dirOut,paste0(file.code,"Monocle_DEA_genes.csv")),row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaMonocle[deaMonocle$cell_group == cl.val,"cell_group"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "cell_group",
replacement = "clusters")
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "gene_id",
replacement = "genes")
deaMonocle$data_set <- subset.datasets_csv[ind,1]
deaMonocle$set_number <- ind
deaMonocle <- as.data.frame(deaMonocle)
deaMonocle_tot <- rbind(deaMonocle_tot, deaMonocle)
}
deaMonocle_tot <- deaMonocle_tot[2:nrow(deaMonocle_tot),]
deaMonocle_tot <- merge.data.frame(deaMonocle_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaMonocle_tot$value <- as.numeric(deaMonocle_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultMonocle <- roc(deaMonocle_tot$value, 1 - deaMonocle_tot$marker_test_p_value)
# Plot the ROC curve
#plot(roc_resultMonocle)ROC from ScamPy
deaScamPy_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaScamPy <- read.csv(file.path(dirOut,paste0(file.code,"ScanPy_DEA_genes.csv")),
row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaScamPy[deaScamPy$clusters == paste0("cl",cl.val),"clusters"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
deaScamPy$data_set <- subset.datasets_csv[ind,1]
deaScamPy$set_number <- ind
deaScamPy_tot <- rbind(deaScamPy_tot, deaScamPy)
}
deaScamPy_tot <- deaScamPy_tot[2:nrow(deaScamPy_tot),]
deaScamPy_tot <- merge.data.frame(deaScamPy_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaScamPy_tot$value <- as.numeric(deaScamPy_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultScamPy <- roc(deaScamPy_tot$value, 1 - deaScamPy_tot$pval)
# Plot the ROC curve
#plot(roc_resultScamPy)Summary ROC for all methods
TwoClusters_even_medium <- ggroc(list(COTAN=roc_resultCOTAN, Seurat=roc_resultSeurat,
Seurat_scTr = roc_resultSeurat_scTr, Seurat_Bimod = roc_resultSeurat_Bimod,
Monocle=roc_resultMonocle, ScamPy=roc_resultScamPy))
TwoClusters_even_mediumPL <- TwoClusters_even_medium + xlab("FPR") + ylab("TPR")
TwoClusters_even_mediumPL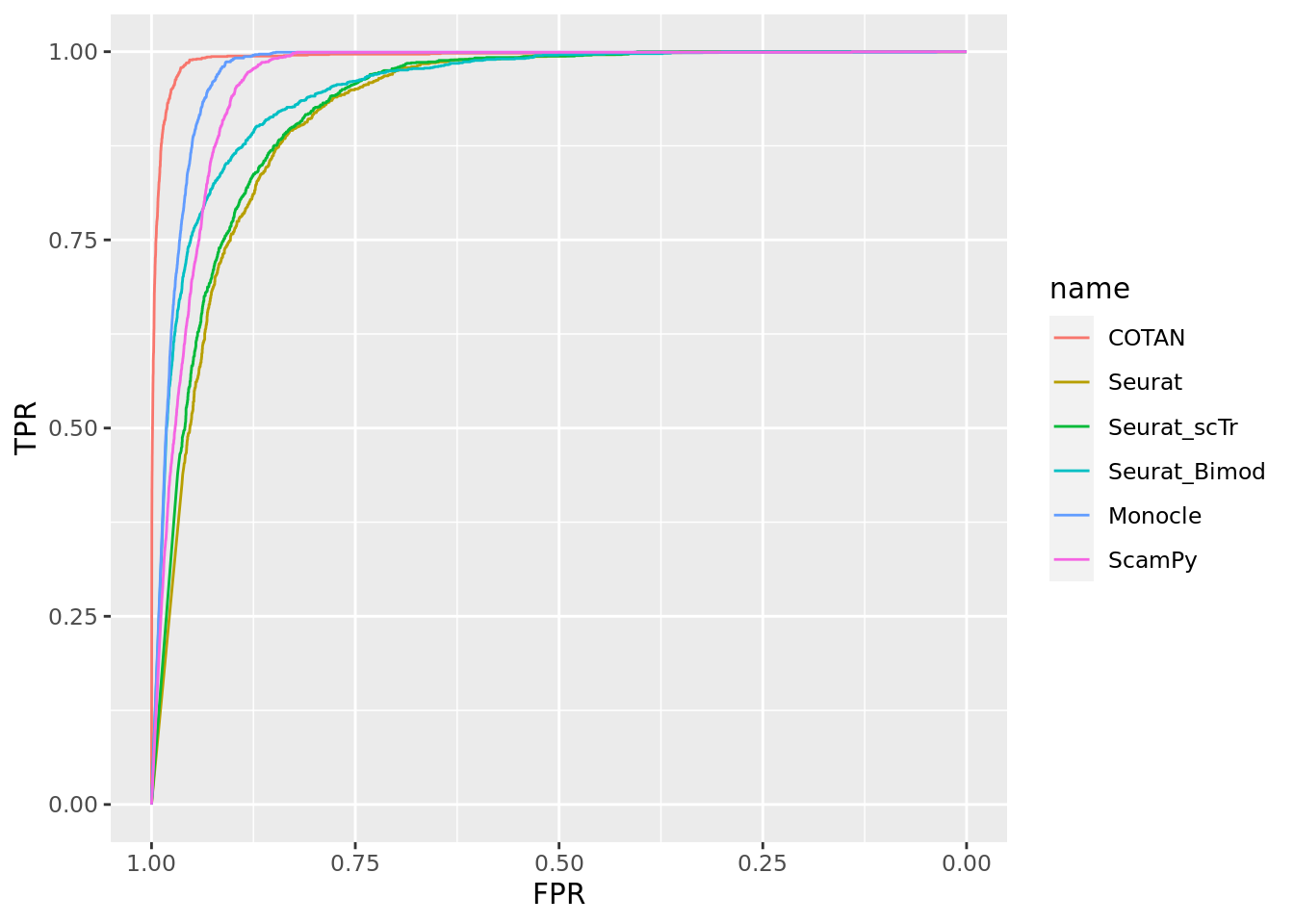
2_Clusters_even_far
True vector
subset.datasets_csv <-datasets_csv[datasets_csv$Group == "2_Clusters_even_far", ]
ground_truth_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
clusters <- str_split(subset.datasets_csv$Collection[ind], pattern = "_[+]_", simplify = T)
reads.LM.subset <- readsLogMeansPerCluster[, clusters]
ground_truth <- as.data.frame(matrix(nrow = nrow(reads.LM.subset),
ncol = ncol(reads.LM.subset)))
rownames(ground_truth) <- rownames(reads.LM.subset)
colnames(ground_truth) <- colnames(reads.LM.subset)
for (col in 1:ncol(ground_truth)) {
# log fold change
ground_truth[, col] <- reads.LM.subset[, col] - rowMeans(reads.LM.subset[, -col, drop = FALSE])
ground_truth[, col] <- ground_truth[, col] > thresholdLFC & reads.LM.subset[, col] > thresholdLFM
}
ground_truth$genes <- rownames(ground_truth)
ground_truth <- pivot_longer(ground_truth,
cols = 1:(ncol(ground_truth)-1),
names_to = "clusters")
ground_truth$data_set <- subset.datasets_csv[ind, 1]
ground_truth$set_number <- ind
ground_truth_tot <- rbind(ground_truth_tot, ground_truth)
}
ground_truth_tot <- ground_truth_tot[2:nrow(ground_truth_tot),]
head(ground_truth_tot)# A tibble: 6 × 5
genes clusters value data_set set_number
<chr> <chr> <lgl> <chr> <int>
1 Neil2 E17.5-516 FALSE 2_Clusters_even_far 1
2 Neil2 E13.5-187 FALSE 2_Clusters_even_far 1
3 Lamc1 E17.5-516 FALSE 2_Clusters_even_far 1
4 Lamc1 E13.5-187 FALSE 2_Clusters_even_far 1
5 Lama1 E17.5-516 FALSE 2_Clusters_even_far 1
6 Lama1 E13.5-187 FALSE 2_Clusters_even_far 1length(unique(ground_truth_tot$genes))[1] 14695sum(ground_truth_tot$value)[1] 4020ROC for COTAN
onlyPositive.pVal.Cotan_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",
subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
deaCOTAN <- getClusterizationData(dataset,clName = "mergedClusters")[[2]]
pvalCOTAN <- pValueFromDEA(deaCOTAN,
numCells = getNumCells(dataset),adjustmentMethod = "none")
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.names <- c(cl.names,
str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1])
cl.names <- cl.names[!is.na(cl.names)]
}
colnames(deaCOTAN) <- cl.names
colnames(pvalCOTAN) <- cl.names
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
onlyPositive.pVal.Cotan <- pvalCOTAN
for (cl in cl.names) {
print(cl)
#temp.DEA.CotanSign <- deaCOTAN[rownames(pvalCOTAN[pvalCOTAN[,cl] < 0.05,]) ,]
onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl] <- 1 #onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl]+1
}
onlyPositive.pVal.Cotan$genes <- rownames(onlyPositive.pVal.Cotan)
onlyPositive.pVal.Cotan <- pivot_longer(onlyPositive.pVal.Cotan,
values_to = "p_values",
cols = 1:(ncol(onlyPositive.pVal.Cotan)-1),
names_to = "clusters")
onlyPositive.pVal.Cotan$data_set <- subset.datasets_csv[ind,1]
onlyPositive.pVal.Cotan$set_number <- ind
onlyPositive.pVal.Cotan_tot <- rbind(onlyPositive.pVal.Cotan_tot, onlyPositive.pVal.Cotan)
}[1] "E13.5-187"
[1] "E17.5-516"
[1] "E15.0-510"
[1] "E13.5-437"
[1] "E15.0-509"
[1] "E13.5-184"onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[2:nrow(onlyPositive.pVal.Cotan_tot),]
onlyPositive.pVal.Cotan_tot <- merge.data.frame(onlyPositive.pVal.Cotan_tot,
ground_truth_tot,by = c("genes","clusters","data_set","set_number"),all.x = T,all.y = F)
#onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[order(onlyPositive.pVal.Cotan_tot$p_values,
# decreasing = F),]
# df <- as.data.frame(matrix(nrow = nrow(onlyPositive.pVal.Cotan_tot),ncol = 3))
# colnames(df) <- c("TPR","FPR","Method")
# df$Method <- "COTAN"
#
# Positive <- sum(onlyPositive.pVal.Cotan_tot$value)
# Negative <- sum(!onlyPositive.pVal.Cotan_tot$value)
#
# for (i in 1:nrow(onlyPositive.pVal.Cotan_tot)) {
# df[i,"TPR"] <- sum(onlyPositive.pVal.Cotan_tot[1:i,"value"])/Positive
# df[i,"FPR"] <- (i-sum(onlyPositive.pVal.Cotan_tot[1:i,"value"]))/Negative
# Convert TRUE/FALSE to 1/0
onlyPositive.pVal.Cotan_tot$value <- as.numeric(onlyPositive.pVal.Cotan_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultCOTAN <- roc(onlyPositive.pVal.Cotan_tot$value, 1 - onlyPositive.pVal.Cotan_tot$p_values)
# Plot the ROC curve
#plot(roc_resultCOTAN)ROC for Seurat
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat scTransform
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_ScTransform_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_scTr <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat Bimod
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_Bimod_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_Bimod <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Monocle
deaMonocle_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaMonocle <- read.csv(file.path(dirOut,paste0(file.code,"Monocle_DEA_genes.csv")),row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaMonocle[deaMonocle$cell_group == cl.val,"cell_group"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "cell_group",
replacement = "clusters")
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "gene_id",
replacement = "genes")
deaMonocle$data_set <- subset.datasets_csv[ind,1]
deaMonocle$set_number <- ind
deaMonocle <- as.data.frame(deaMonocle)
deaMonocle_tot <- rbind(deaMonocle_tot, deaMonocle)
}
deaMonocle_tot <- deaMonocle_tot[2:nrow(deaMonocle_tot),]
deaMonocle_tot <- merge.data.frame(deaMonocle_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaMonocle_tot$value <- as.numeric(deaMonocle_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultMonocle <- roc(deaMonocle_tot$value, 1 - deaMonocle_tot$marker_test_p_value)
# Plot the ROC curve
#plot(roc_resultMonocle)ROC from ScamPy
deaScamPy_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaScamPy <- read.csv(file.path(dirOut,paste0(file.code,"ScanPy_DEA_genes.csv")),
row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaScamPy[deaScamPy$clusters == paste0("cl",cl.val),"clusters"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
deaScamPy$data_set <- subset.datasets_csv[ind,1]
deaScamPy$set_number <- ind
deaScamPy_tot <- rbind(deaScamPy_tot, deaScamPy)
}
deaScamPy_tot <- deaScamPy_tot[2:nrow(deaScamPy_tot),]
deaScamPy_tot <- merge.data.frame(deaScamPy_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaScamPy_tot$value <- as.numeric(deaScamPy_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultScamPy <- roc(deaScamPy_tot$value, 1 - deaScamPy_tot$pval)
# Plot the ROC curve
#plot(roc_resultScamPy)Summary ROC for all methods
TwoClusters_even_far <- ggroc(list(COTAN=roc_resultCOTAN, Seurat=roc_resultSeurat,
Seurat_scTr = roc_resultSeurat_scTr, Seurat_Bimod = roc_resultSeurat_Bimod,
Monocle=roc_resultMonocle, ScamPy=roc_resultScamPy))
TwoClusters_even_farPL <- TwoClusters_even_far + xlab("FPR") + ylab("TPR")
TwoClusters_even_farPL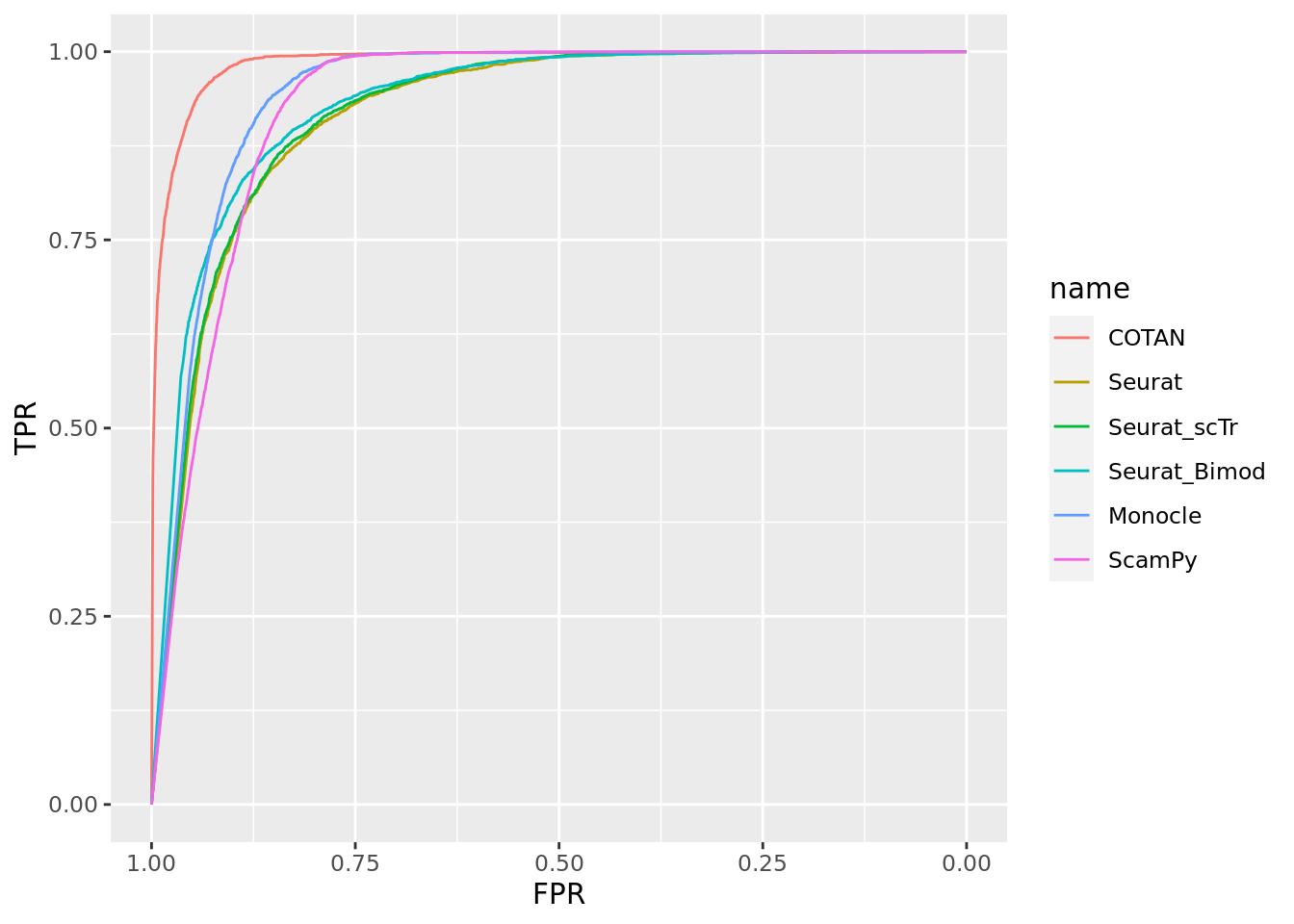
2 Clusters uneven
2_Clusters_uneven_near
True vector
subset.datasets_csv <-datasets_csv[datasets_csv$Group == "2_Clusters_uneven_near", ]
ground_truth_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
clusters <- str_split(subset.datasets_csv$Collection[ind], pattern = "_[+]_", simplify = T)
reads.LM.subset <- readsLogMeansPerCluster[, clusters]
ground_truth <- as.data.frame(matrix(nrow = nrow(reads.LM.subset),
ncol = ncol(reads.LM.subset)))
rownames(ground_truth) <- rownames(reads.LM.subset)
colnames(ground_truth) <- colnames(reads.LM.subset)
for (col in 1:ncol(ground_truth)) {
# log fold change
ground_truth[, col] <- reads.LM.subset[, col] - rowMeans(reads.LM.subset[, -col, drop = FALSE])
ground_truth[, col] <- ground_truth[, col] > thresholdLFC & reads.LM.subset[, col] > thresholdLFM
}
ground_truth$genes <- rownames(ground_truth)
ground_truth <- pivot_longer(ground_truth,
cols = 1:(ncol(ground_truth)-1),
names_to = "clusters")
ground_truth$data_set <- subset.datasets_csv[ind, 1]
ground_truth$set_number <- ind
ground_truth_tot <- rbind(ground_truth_tot, ground_truth)
}
ground_truth_tot <- ground_truth_tot[2:nrow(ground_truth_tot),]
head(ground_truth_tot)# A tibble: 6 × 5
genes clusters value data_set set_number
<chr> <chr> <lgl> <chr> <int>
1 Neil2 E13.5-434 FALSE 2_Clusters_uneven_near 1
2 Neil2 E15.0-428 FALSE 2_Clusters_uneven_near 1
3 Lamc1 E13.5-434 FALSE 2_Clusters_uneven_near 1
4 Lamc1 E15.0-428 FALSE 2_Clusters_uneven_near 1
5 Lama1 E13.5-434 FALSE 2_Clusters_uneven_near 1
6 Lama1 E15.0-428 FALSE 2_Clusters_uneven_near 1length(unique(ground_truth_tot$genes))[1] 14695sum(ground_truth_tot$value)[1] 80ROC for COTAN
onlyPositive.pVal.Cotan_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",
subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
deaCOTAN <- getClusterizationData(dataset,clName = "mergedClusters")[[2]]
pvalCOTAN <- pValueFromDEA(deaCOTAN,
numCells = getNumCells(dataset),adjustmentMethod = "none")
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.names <- c(cl.names,
str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1])
cl.names <- cl.names[!is.na(cl.names)]
}
colnames(deaCOTAN) <- cl.names
colnames(pvalCOTAN) <- cl.names
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
onlyPositive.pVal.Cotan <- pvalCOTAN
for (cl in cl.names) {
print(cl)
#temp.DEA.CotanSign <- deaCOTAN[rownames(pvalCOTAN[pvalCOTAN[,cl] < 0.05,]) ,]
onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl] <- 1 #onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl]+1
}
onlyPositive.pVal.Cotan$genes <- rownames(onlyPositive.pVal.Cotan)
onlyPositive.pVal.Cotan <- pivot_longer(onlyPositive.pVal.Cotan,
values_to = "p_values",
cols = 1:(ncol(onlyPositive.pVal.Cotan)-1),
names_to = "clusters")
onlyPositive.pVal.Cotan$data_set <- subset.datasets_csv[ind,1]
onlyPositive.pVal.Cotan$set_number <- ind
onlyPositive.pVal.Cotan_tot <- rbind(onlyPositive.pVal.Cotan_tot, onlyPositive.pVal.Cotan)
}[1] "E13.5-434"
[1] "E15.0-428"
[1] "E15.0-432"
[1] "E13.5-432"
[1] "E15.0-509"
[1] "E15.0-508"onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[2:nrow(onlyPositive.pVal.Cotan_tot),]
onlyPositive.pVal.Cotan_tot <- merge.data.frame(onlyPositive.pVal.Cotan_tot,
ground_truth_tot,by = c("genes","clusters","data_set","set_number"),all.x = T,all.y = F)
#onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[order(onlyPositive.pVal.Cotan_tot$p_values,
# decreasing = F),]
# df <- as.data.frame(matrix(nrow = nrow(onlyPositive.pVal.Cotan_tot),ncol = 3))
# colnames(df) <- c("TPR","FPR","Method")
# df$Method <- "COTAN"
#
# Positive <- sum(onlyPositive.pVal.Cotan_tot$value)
# Negative <- sum(!onlyPositive.pVal.Cotan_tot$value)
#
# for (i in 1:nrow(onlyPositive.pVal.Cotan_tot)) {
# df[i,"TPR"] <- sum(onlyPositive.pVal.Cotan_tot[1:i,"value"])/Positive
# df[i,"FPR"] <- (i-sum(onlyPositive.pVal.Cotan_tot[1:i,"value"]))/Negative
# Convert TRUE/FALSE to 1/0
onlyPositive.pVal.Cotan_tot$value <- as.numeric(onlyPositive.pVal.Cotan_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultCOTAN <- roc(onlyPositive.pVal.Cotan_tot$value, 1 - onlyPositive.pVal.Cotan_tot$p_values)
# Plot the ROC curve
#plot(roc_resultCOTAN)ROC for Seurat
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat scTransform
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_ScTransform_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_scTr <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat Bimod
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_Bimod_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_Bimod <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Monocle
deaMonocle_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaMonocle <- read.csv(file.path(dirOut,paste0(file.code,"Monocle_DEA_genes.csv")),row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaMonocle[deaMonocle$cell_group == cl.val,"cell_group"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "cell_group",
replacement = "clusters")
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "gene_id",
replacement = "genes")
deaMonocle$data_set <- subset.datasets_csv[ind,1]
deaMonocle$set_number <- ind
deaMonocle <- as.data.frame(deaMonocle)
deaMonocle_tot <- rbind(deaMonocle_tot, deaMonocle)
}
deaMonocle_tot <- deaMonocle_tot[2:nrow(deaMonocle_tot),]
deaMonocle_tot <- merge.data.frame(deaMonocle_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaMonocle_tot$value <- as.numeric(deaMonocle_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultMonocle <- roc(deaMonocle_tot$value, 1 - deaMonocle_tot$marker_test_p_value)
# Plot the ROC curve
#plot(roc_resultMonocle)ROC from ScamPy
deaScamPy_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaScamPy <- read.csv(file.path(dirOut,paste0(file.code,"ScanPy_DEA_genes.csv")),
row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaScamPy[deaScamPy$clusters == paste0("cl",cl.val),"clusters"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
deaScamPy$data_set <- subset.datasets_csv[ind,1]
deaScamPy$set_number <- ind
deaScamPy_tot <- rbind(deaScamPy_tot, deaScamPy)
}
deaScamPy_tot <- deaScamPy_tot[2:nrow(deaScamPy_tot),]
deaScamPy_tot <- merge.data.frame(deaScamPy_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaScamPy_tot$value <- as.numeric(deaScamPy_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultScamPy <- roc(deaScamPy_tot$value, 1 - deaScamPy_tot$pval)
# Plot the ROC curve
#plot(roc_resultScamPy)Summary ROC for all methods
TwoClusters_uneven_near <- ggroc(list(COTAN=roc_resultCOTAN, Seurat=roc_resultSeurat,
Seurat_scTr = roc_resultSeurat_scTr, Seurat_Bimod = roc_resultSeurat_Bimod,
Monocle=roc_resultMonocle, ScamPy=roc_resultScamPy))
TwoClusters_uneven_nearPL <- TwoClusters_uneven_near + xlab("FPR") + ylab("TPR")
TwoClusters_uneven_nearPL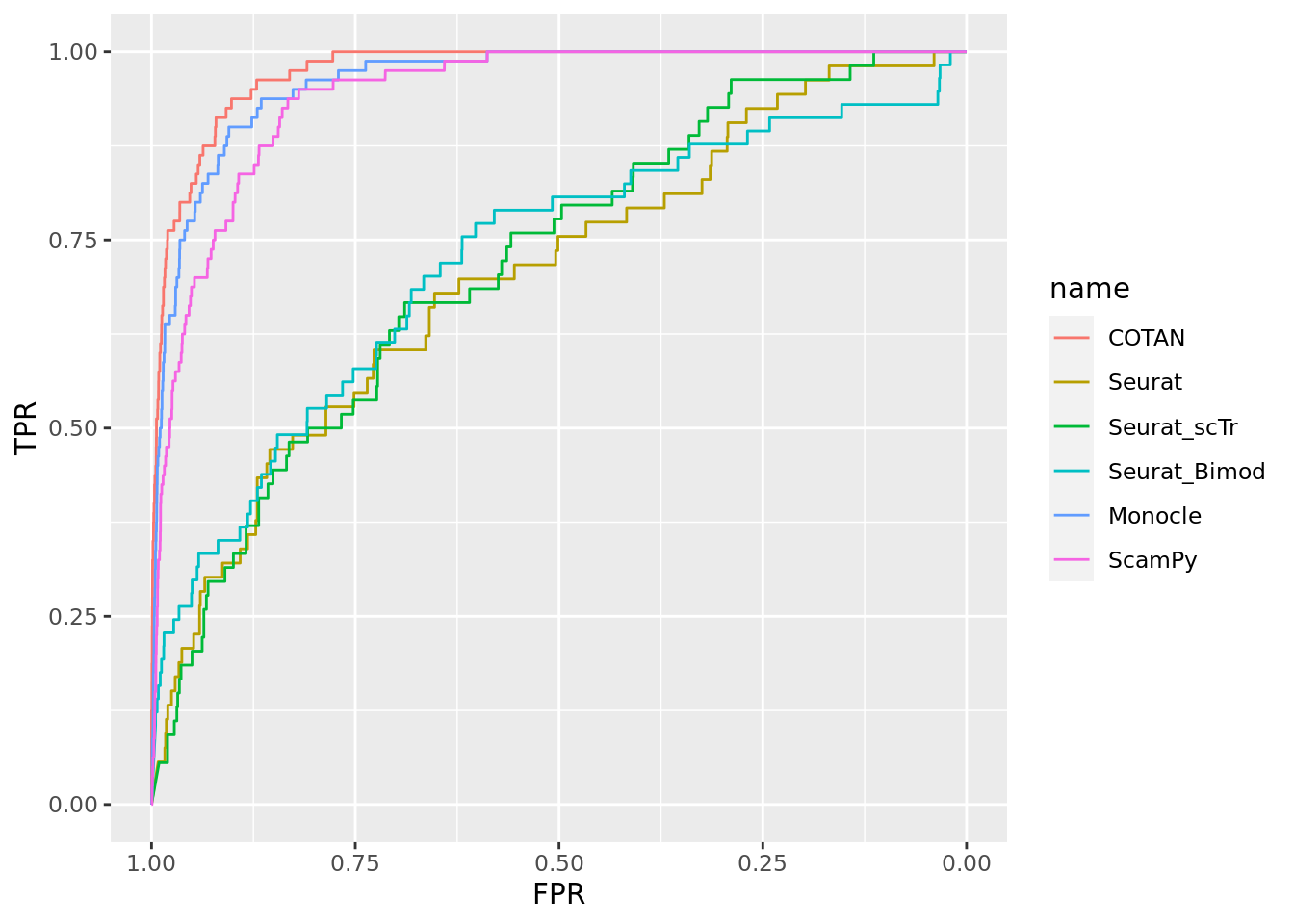
2_Clusters_uneven_medium
True vector
subset.datasets_csv <-datasets_csv[datasets_csv$Group == "2_Clusters_uneven_medium", ]
ground_truth_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
clusters <- str_split(subset.datasets_csv$Collection[ind], pattern = "_[+]_", simplify = T)
reads.LM.subset <- readsLogMeansPerCluster[, clusters]
ground_truth <- as.data.frame(matrix(nrow = nrow(reads.LM.subset),
ncol = ncol(reads.LM.subset)))
rownames(ground_truth) <- rownames(reads.LM.subset)
colnames(ground_truth) <- colnames(reads.LM.subset)
for (col in 1:ncol(ground_truth)) {
# log fold change
ground_truth[, col] <- reads.LM.subset[, col] - rowMeans(reads.LM.subset[, -col, drop = FALSE])
ground_truth[, col] <- ground_truth[, col] > thresholdLFC & reads.LM.subset[, col] > thresholdLFM
}
ground_truth$genes <- rownames(ground_truth)
ground_truth <- pivot_longer(ground_truth,
cols = 1:(ncol(ground_truth)-1),
names_to = "clusters")
ground_truth$data_set <- subset.datasets_csv[ind, 1]
ground_truth$set_number <- ind
ground_truth_tot <- rbind(ground_truth_tot, ground_truth)
}
ground_truth_tot <- ground_truth_tot[2:nrow(ground_truth_tot),]
head(ground_truth_tot)# A tibble: 6 × 5
genes clusters value data_set set_number
<chr> <chr> <lgl> <chr> <int>
1 Neil2 E13.5-187 FALSE 2_Clusters_uneven_medium 1
2 Neil2 E13.5-184 FALSE 2_Clusters_uneven_medium 1
3 Lamc1 E13.5-187 FALSE 2_Clusters_uneven_medium 1
4 Lamc1 E13.5-184 FALSE 2_Clusters_uneven_medium 1
5 Lama1 E13.5-187 FALSE 2_Clusters_uneven_medium 1
6 Lama1 E13.5-184 FALSE 2_Clusters_uneven_medium 1length(unique(ground_truth_tot$genes))[1] 14695sum(ground_truth_tot$value)[1] 1559ROC for COTAN
onlyPositive.pVal.Cotan_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",
subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
deaCOTAN <- getClusterizationData(dataset,clName = "mergedClusters")[[2]]
pvalCOTAN <- pValueFromDEA(deaCOTAN,
numCells = getNumCells(dataset),adjustmentMethod = "none")
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.names <- c(cl.names,
str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1])
cl.names <- cl.names[!is.na(cl.names)]
}
colnames(deaCOTAN) <- cl.names
colnames(pvalCOTAN) <- cl.names
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
onlyPositive.pVal.Cotan <- pvalCOTAN
for (cl in cl.names) {
print(cl)
#temp.DEA.CotanSign <- deaCOTAN[rownames(pvalCOTAN[pvalCOTAN[,cl] < 0.05,]) ,]
onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl] <- 1 #onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl]+1
}
onlyPositive.pVal.Cotan$genes <- rownames(onlyPositive.pVal.Cotan)
onlyPositive.pVal.Cotan <- pivot_longer(onlyPositive.pVal.Cotan,
values_to = "p_values",
cols = 1:(ncol(onlyPositive.pVal.Cotan)-1),
names_to = "clusters")
onlyPositive.pVal.Cotan$data_set <- subset.datasets_csv[ind,1]
onlyPositive.pVal.Cotan$set_number <- ind
onlyPositive.pVal.Cotan_tot <- rbind(onlyPositive.pVal.Cotan_tot, onlyPositive.pVal.Cotan)
}[1] "E13.5-187"
[1] "E13.5-184"
[1] "E17.5-516"
[1] "E15.0-434"
[1] "E15.0-508"
[1] "E15.0-437"onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[2:nrow(onlyPositive.pVal.Cotan_tot),]
onlyPositive.pVal.Cotan_tot <- merge.data.frame(onlyPositive.pVal.Cotan_tot,
ground_truth_tot,by = c("genes","clusters","data_set","set_number"),all.x = T,all.y = F)
#onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[order(onlyPositive.pVal.Cotan_tot$p_values,
# decreasing = F),]
# df <- as.data.frame(matrix(nrow = nrow(onlyPositive.pVal.Cotan_tot),ncol = 3))
# colnames(df) <- c("TPR","FPR","Method")
# df$Method <- "COTAN"
#
# Positive <- sum(onlyPositive.pVal.Cotan_tot$value)
# Negative <- sum(!onlyPositive.pVal.Cotan_tot$value)
#
# for (i in 1:nrow(onlyPositive.pVal.Cotan_tot)) {
# df[i,"TPR"] <- sum(onlyPositive.pVal.Cotan_tot[1:i,"value"])/Positive
# df[i,"FPR"] <- (i-sum(onlyPositive.pVal.Cotan_tot[1:i,"value"]))/Negative
# Convert TRUE/FALSE to 1/0
onlyPositive.pVal.Cotan_tot$value <- as.numeric(onlyPositive.pVal.Cotan_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultCOTAN <- roc(onlyPositive.pVal.Cotan_tot$value, 1 - onlyPositive.pVal.Cotan_tot$p_values)
# Plot the ROC curve
#plot(roc_resultCOTAN)ROC for Seurat
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat scTransform
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_ScTransform_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_scTr <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat Bimod
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_Bimod_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_Bimod <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Monocle
deaMonocle_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaMonocle <- read.csv(file.path(dirOut,paste0(file.code,"Monocle_DEA_genes.csv")),row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaMonocle[deaMonocle$cell_group == cl.val,"cell_group"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "cell_group",
replacement = "clusters")
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "gene_id",
replacement = "genes")
deaMonocle$data_set <- subset.datasets_csv[ind,1]
deaMonocle$set_number <- ind
deaMonocle <- as.data.frame(deaMonocle)
deaMonocle_tot <- rbind(deaMonocle_tot, deaMonocle)
}
deaMonocle_tot <- deaMonocle_tot[2:nrow(deaMonocle_tot),]
deaMonocle_tot <- merge.data.frame(deaMonocle_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaMonocle_tot$value <- as.numeric(deaMonocle_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultMonocle <- roc(deaMonocle_tot$value, 1 - deaMonocle_tot$marker_test_p_value)
# Plot the ROC curve
#plot(roc_resultMonocle)ROC from ScamPy
deaScamPy_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaScamPy <- read.csv(file.path(dirOut,paste0(file.code,"ScanPy_DEA_genes.csv")),
row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaScamPy[deaScamPy$clusters == paste0("cl",cl.val),"clusters"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
deaScamPy$data_set <- subset.datasets_csv[ind,1]
deaScamPy$set_number <- ind
deaScamPy_tot <- rbind(deaScamPy_tot, deaScamPy)
}
deaScamPy_tot <- deaScamPy_tot[2:nrow(deaScamPy_tot),]
deaScamPy_tot <- merge.data.frame(deaScamPy_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaScamPy_tot$value <- as.numeric(deaScamPy_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultScamPy <- roc(deaScamPy_tot$value, 1 - deaScamPy_tot$pval)
# Plot the ROC curve
#plot(roc_resultScamPy)Summary ROC for all methods
TwoClusters_uneven_medium <- ggroc(list(COTAN=roc_resultCOTAN, Seurat=roc_resultSeurat,
Seurat_scTr = roc_resultSeurat_scTr, Seurat_Bimod = roc_resultSeurat_Bimod,
Monocle=roc_resultMonocle, ScamPy=roc_resultScamPy))
TwoClusters_uneven_mediumPL <- TwoClusters_uneven_medium + xlab("FPR") + ylab("TPR")
TwoClusters_uneven_mediumPL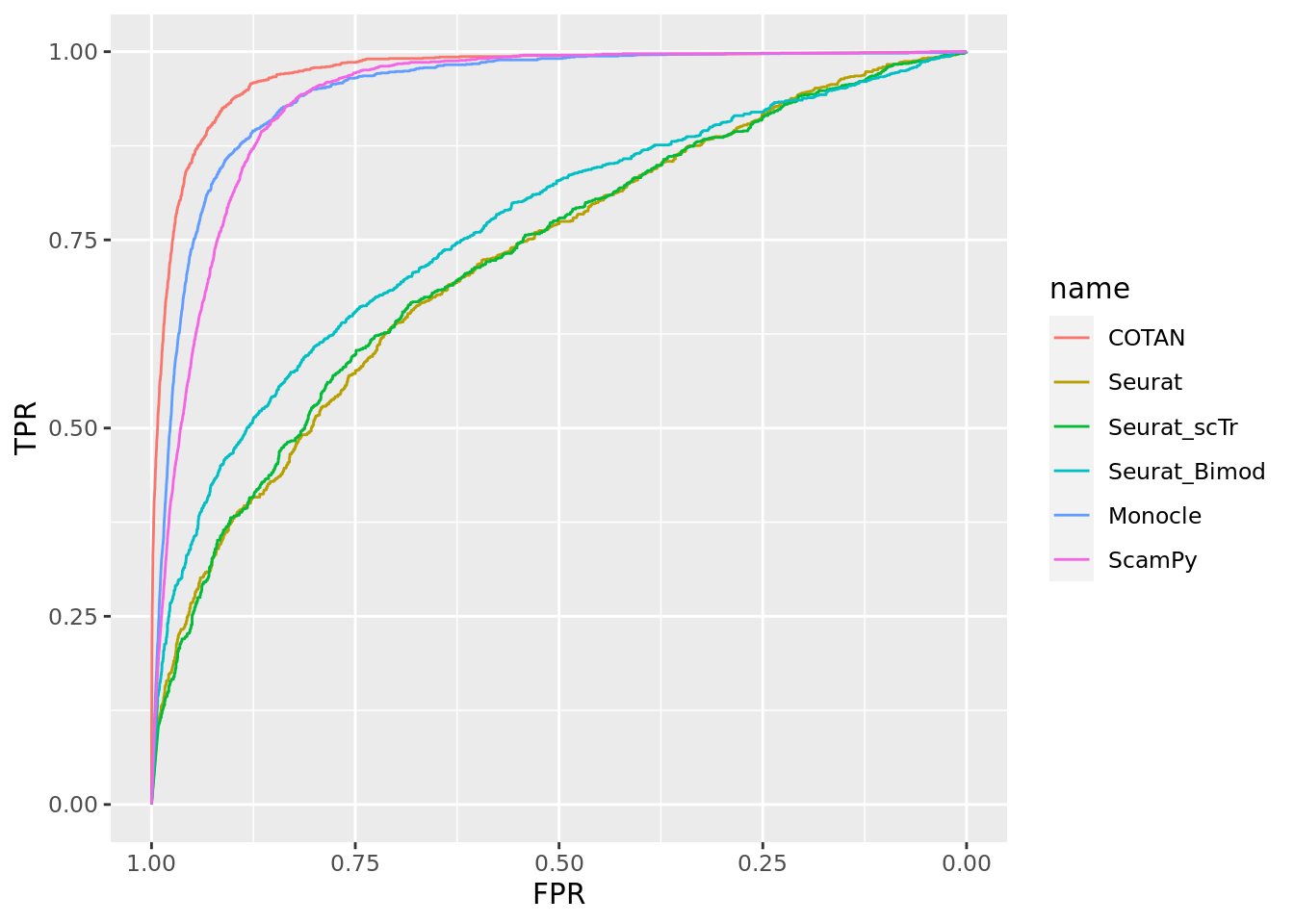
2_Clusters_uneven_far
True vector
subset.datasets_csv <-datasets_csv[datasets_csv$Group == "2_Clusters_uneven_far", ]
ground_truth_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
clusters <- str_split(subset.datasets_csv$Collection[ind], pattern = "_[+]_", simplify = T)
reads.LM.subset <- readsLogMeansPerCluster[, clusters]
ground_truth <- as.data.frame(matrix(nrow = nrow(reads.LM.subset),
ncol = ncol(reads.LM.subset)))
rownames(ground_truth) <- rownames(reads.LM.subset)
colnames(ground_truth) <- colnames(reads.LM.subset)
for (col in 1:ncol(ground_truth)) {
# log fold change
ground_truth[, col] <- reads.LM.subset[, col] - rowMeans(reads.LM.subset[, -col, drop = FALSE])
ground_truth[, col] <- ground_truth[, col] > thresholdLFC & reads.LM.subset[, col] > thresholdLFM
}
ground_truth$genes <- rownames(ground_truth)
ground_truth <- pivot_longer(ground_truth,
cols = 1:(ncol(ground_truth)-1),
names_to = "clusters")
ground_truth$data_set <- subset.datasets_csv[ind, 1]
ground_truth$set_number <- ind
ground_truth_tot <- rbind(ground_truth_tot, ground_truth)
}
ground_truth_tot <- ground_truth_tot[2:nrow(ground_truth_tot),]
head(ground_truth_tot)# A tibble: 6 × 5
genes clusters value data_set set_number
<chr> <chr> <lgl> <chr> <int>
1 Neil2 E17.5-516 FALSE 2_Clusters_uneven_far 1
2 Neil2 E13.5-187 FALSE 2_Clusters_uneven_far 1
3 Lamc1 E17.5-516 FALSE 2_Clusters_uneven_far 1
4 Lamc1 E13.5-187 FALSE 2_Clusters_uneven_far 1
5 Lama1 E17.5-516 FALSE 2_Clusters_uneven_far 1
6 Lama1 E13.5-187 FALSE 2_Clusters_uneven_far 1length(unique(ground_truth_tot$genes))[1] 14695sum(ground_truth_tot$value)[1] 4020ROC for COTAN
onlyPositive.pVal.Cotan_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",
subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
deaCOTAN <- getClusterizationData(dataset,clName = "mergedClusters")[[2]]
pvalCOTAN <- pValueFromDEA(deaCOTAN,
numCells = getNumCells(dataset),adjustmentMethod = "none")
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.names <- c(cl.names,
str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1])
cl.names <- cl.names[!is.na(cl.names)]
}
colnames(deaCOTAN) <- cl.names
colnames(pvalCOTAN) <- cl.names
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
onlyPositive.pVal.Cotan <- pvalCOTAN
for (cl in cl.names) {
print(cl)
#temp.DEA.CotanSign <- deaCOTAN[rownames(pvalCOTAN[pvalCOTAN[,cl] < 0.05,]) ,]
onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl] <- 1 #onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl]+1
}
onlyPositive.pVal.Cotan$genes <- rownames(onlyPositive.pVal.Cotan)
onlyPositive.pVal.Cotan <- pivot_longer(onlyPositive.pVal.Cotan,
values_to = "p_values",
cols = 1:(ncol(onlyPositive.pVal.Cotan)-1),
names_to = "clusters")
onlyPositive.pVal.Cotan$data_set <- subset.datasets_csv[ind,1]
onlyPositive.pVal.Cotan$set_number <- ind
onlyPositive.pVal.Cotan_tot <- rbind(onlyPositive.pVal.Cotan_tot, onlyPositive.pVal.Cotan)
}[1] "E13.5-187"
[1] "E17.5-516"
[1] "E15.0-510"
[1] "E13.5-437"
[1] "E15.0-509"
[1] "E13.5-184"onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[2:nrow(onlyPositive.pVal.Cotan_tot),]
onlyPositive.pVal.Cotan_tot <- merge.data.frame(onlyPositive.pVal.Cotan_tot,
ground_truth_tot,by = c("genes","clusters","data_set","set_number"),all.x = T,all.y = F)
#onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[order(onlyPositive.pVal.Cotan_tot$p_values,
# decreasing = F),]
# df <- as.data.frame(matrix(nrow = nrow(onlyPositive.pVal.Cotan_tot),ncol = 3))
# colnames(df) <- c("TPR","FPR","Method")
# df$Method <- "COTAN"
#
# Positive <- sum(onlyPositive.pVal.Cotan_tot$value)
# Negative <- sum(!onlyPositive.pVal.Cotan_tot$value)
#
# for (i in 1:nrow(onlyPositive.pVal.Cotan_tot)) {
# df[i,"TPR"] <- sum(onlyPositive.pVal.Cotan_tot[1:i,"value"])/Positive
# df[i,"FPR"] <- (i-sum(onlyPositive.pVal.Cotan_tot[1:i,"value"]))/Negative
# Convert TRUE/FALSE to 1/0
onlyPositive.pVal.Cotan_tot$value <- as.numeric(onlyPositive.pVal.Cotan_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultCOTAN <- roc(onlyPositive.pVal.Cotan_tot$value, 1 - onlyPositive.pVal.Cotan_tot$p_values)
# Plot the ROC curve
#plot(roc_resultCOTAN)ROC for Seurat
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat scTransform
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_ScTransform_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_scTr <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat Bimod
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_Bimod_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_Bimod <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Monocle
deaMonocle_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaMonocle <- read.csv(file.path(dirOut,paste0(file.code,"Monocle_DEA_genes.csv")),row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaMonocle[deaMonocle$cell_group == cl.val,"cell_group"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "cell_group",
replacement = "clusters")
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "gene_id",
replacement = "genes")
deaMonocle$data_set <- subset.datasets_csv[ind,1]
deaMonocle$set_number <- ind
deaMonocle <- as.data.frame(deaMonocle)
deaMonocle_tot <- rbind(deaMonocle_tot, deaMonocle)
}
deaMonocle_tot <- deaMonocle_tot[2:nrow(deaMonocle_tot),]
deaMonocle_tot <- merge.data.frame(deaMonocle_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaMonocle_tot$value <- as.numeric(deaMonocle_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultMonocle <- roc(deaMonocle_tot$value, 1 - deaMonocle_tot$marker_test_p_value)
# Plot the ROC curve
#plot(roc_resultMonocle)ROC from ScamPy
deaScamPy_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaScamPy <- read.csv(file.path(dirOut,paste0(file.code,"ScanPy_DEA_genes.csv")),
row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaScamPy[deaScamPy$clusters == paste0("cl",cl.val),"clusters"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
deaScamPy$data_set <- subset.datasets_csv[ind,1]
deaScamPy$set_number <- ind
deaScamPy_tot <- rbind(deaScamPy_tot, deaScamPy)
}
deaScamPy_tot <- deaScamPy_tot[2:nrow(deaScamPy_tot),]
deaScamPy_tot <- merge.data.frame(deaScamPy_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaScamPy_tot$value <- as.numeric(deaScamPy_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultScamPy <- roc(deaScamPy_tot$value, 1 - deaScamPy_tot$pval)
# Plot the ROC curve
#plot(roc_resultScamPy)Summary ROC for all methods
TwoClusters_uneven_far <- ggroc(list(COTAN=roc_resultCOTAN, Seurat=roc_resultSeurat,
Seurat_scTr = roc_resultSeurat_scTr, Seurat_Bimod = roc_resultSeurat_Bimod,
Monocle=roc_resultMonocle, ScamPy=roc_resultScamPy))
TwoClusters_uneven_farPL <- TwoClusters_uneven_far + xlab("FPR") + ylab("TPR")
TwoClusters_uneven_farPL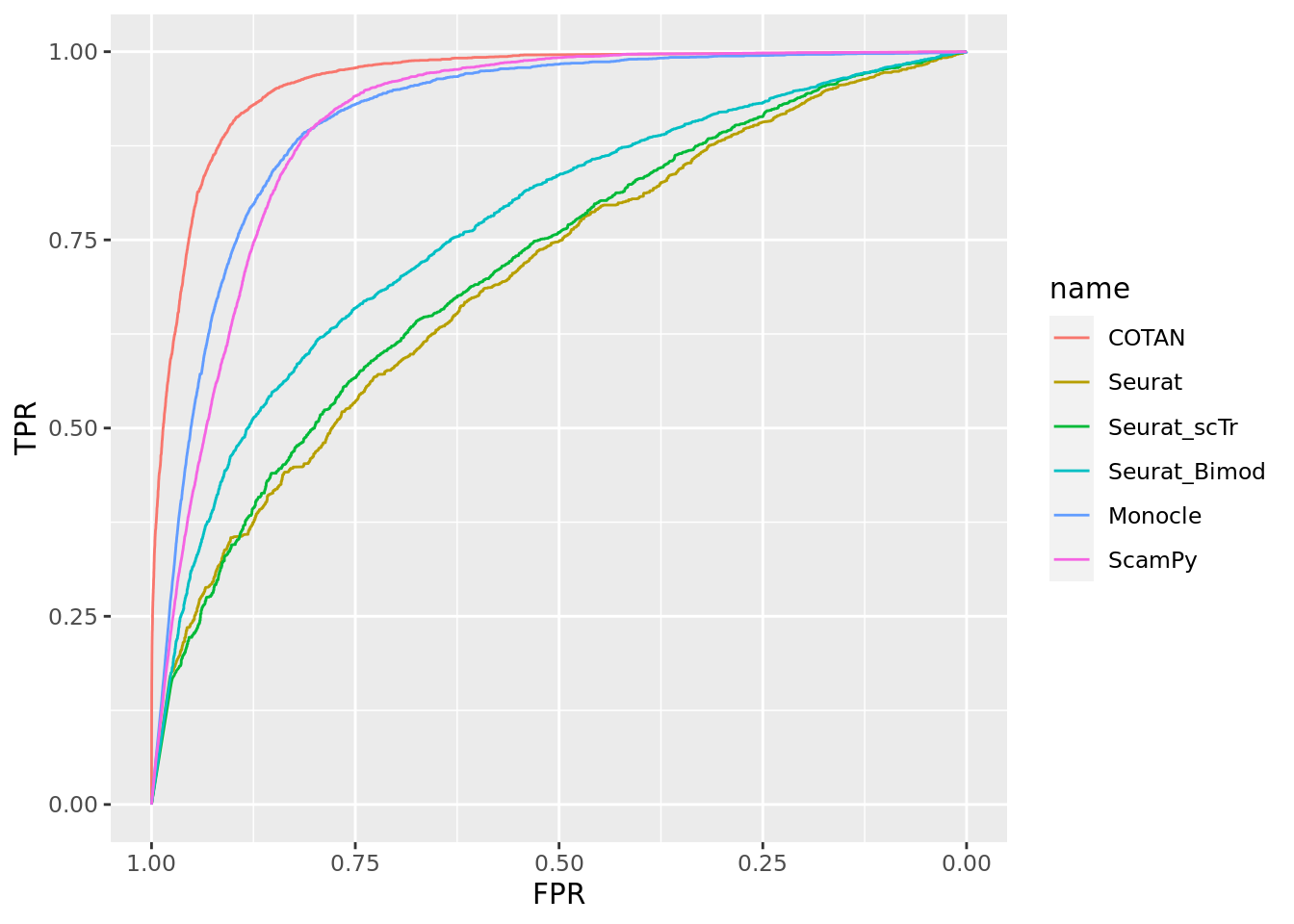
3 clusters
3_Clusters_even
True vector
subset.datasets_csv <-datasets_csv[datasets_csv$Group == "3_Clusters_even", ]
ground_truth_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
clusters <- str_split(subset.datasets_csv$Collection[ind], pattern = "_[+]_", simplify = T)
reads.LM.subset <- readsLogMeansPerCluster[, clusters]
ground_truth <- as.data.frame(matrix(nrow = nrow(reads.LM.subset),
ncol = ncol(reads.LM.subset)))
rownames(ground_truth) <- rownames(reads.LM.subset)
colnames(ground_truth) <- colnames(reads.LM.subset)
for (col in 1:ncol(ground_truth)) {
# log fold change
ground_truth[, col] <- reads.LM.subset[, col] - rowMeans(reads.LM.subset[, -col, drop = FALSE])
ground_truth[, col] <- ground_truth[, col] > thresholdLFC & reads.LM.subset[, col] > thresholdLFM
}
ground_truth$genes <- rownames(ground_truth)
ground_truth <- pivot_longer(ground_truth,
cols = 1:(ncol(ground_truth)-1),
names_to = "clusters")
ground_truth$data_set <- subset.datasets_csv[ind, 1]
ground_truth$set_number <- ind
ground_truth_tot <- rbind(ground_truth_tot, ground_truth)
}
ground_truth_tot <- ground_truth_tot[2:nrow(ground_truth_tot),]
head(ground_truth_tot)# A tibble: 6 × 5
genes clusters value data_set set_number
<chr> <chr> <lgl> <chr> <int>
1 Neil2 E15.0-437 FALSE 3_Clusters_even 1
2 Neil2 E13.5-510 FALSE 3_Clusters_even 1
3 Neil2 E13.5-437 FALSE 3_Clusters_even 1
4 Lamc1 E15.0-437 FALSE 3_Clusters_even 1
5 Lamc1 E13.5-510 FALSE 3_Clusters_even 1
6 Lamc1 E13.5-437 FALSE 3_Clusters_even 1length(unique(ground_truth_tot$genes))[1] 14695sum(ground_truth_tot$value)[1] 2608ROC for COTAN
onlyPositive.pVal.Cotan_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",
subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
deaCOTAN <- getClusterizationData(dataset,clName = "mergedClusters")[[2]]
pvalCOTAN <- pValueFromDEA(deaCOTAN,
numCells = getNumCells(dataset),adjustmentMethod = "none")
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.names <- c(cl.names,
str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1])
cl.names <- cl.names[!is.na(cl.names)]
}
colnames(deaCOTAN) <- cl.names
colnames(pvalCOTAN) <- cl.names
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
onlyPositive.pVal.Cotan <- pvalCOTAN
for (cl in cl.names) {
print(cl)
#temp.DEA.CotanSign <- deaCOTAN[rownames(pvalCOTAN[pvalCOTAN[,cl] < 0.05,]) ,]
onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl] <- 1 #onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl]+1
}
onlyPositive.pVal.Cotan$genes <- rownames(onlyPositive.pVal.Cotan)
onlyPositive.pVal.Cotan <- pivot_longer(onlyPositive.pVal.Cotan,
values_to = "p_values",
cols = 1:(ncol(onlyPositive.pVal.Cotan)-1),
names_to = "clusters")
onlyPositive.pVal.Cotan$data_set <- subset.datasets_csv[ind,1]
onlyPositive.pVal.Cotan$set_number <- ind
onlyPositive.pVal.Cotan_tot <- rbind(onlyPositive.pVal.Cotan_tot, onlyPositive.pVal.Cotan)
}[1] "E13.5-437"
[1] "E15.0-437"
[1] "E13.5-510"
[1] "E17.5-516"
[1] "E13.5-437"
[1] "E17.5-505"
[1] "E15.0-510"
[1] "E15.0-428"
[1] "E13.5-510"onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[2:nrow(onlyPositive.pVal.Cotan_tot),]
onlyPositive.pVal.Cotan_tot <- merge.data.frame(onlyPositive.pVal.Cotan_tot,
ground_truth_tot,by = c("genes","clusters","data_set","set_number"),all.x = T,all.y = F)
#onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[order(onlyPositive.pVal.Cotan_tot$p_values,
# decreasing = F),]
# df <- as.data.frame(matrix(nrow = nrow(onlyPositive.pVal.Cotan_tot),ncol = 3))
# colnames(df) <- c("TPR","FPR","Method")
# df$Method <- "COTAN"
#
# Positive <- sum(onlyPositive.pVal.Cotan_tot$value)
# Negative <- sum(!onlyPositive.pVal.Cotan_tot$value)
#
# for (i in 1:nrow(onlyPositive.pVal.Cotan_tot)) {
# df[i,"TPR"] <- sum(onlyPositive.pVal.Cotan_tot[1:i,"value"])/Positive
# df[i,"FPR"] <- (i-sum(onlyPositive.pVal.Cotan_tot[1:i,"value"]))/Negative
# Convert TRUE/FALSE to 1/0
onlyPositive.pVal.Cotan_tot$value <- as.numeric(onlyPositive.pVal.Cotan_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultCOTAN <- roc(onlyPositive.pVal.Cotan_tot$value, 1 - onlyPositive.pVal.Cotan_tot$p_values)
# Plot the ROC curve
#plot(roc_resultCOTAN)ROC for Seurat
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat scTransform
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_ScTransform_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_scTr <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat Bimod
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_Bimod_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_Bimod <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Monocle
deaMonocle_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaMonocle <- read.csv(file.path(dirOut,paste0(file.code,"Monocle_DEA_genes.csv")),row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaMonocle[deaMonocle$cell_group == cl.val,"cell_group"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "cell_group",
replacement = "clusters")
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "gene_id",
replacement = "genes")
deaMonocle$data_set <- subset.datasets_csv[ind,1]
deaMonocle$set_number <- ind
deaMonocle <- as.data.frame(deaMonocle)
deaMonocle_tot <- rbind(deaMonocle_tot, deaMonocle)
}
deaMonocle_tot <- deaMonocle_tot[2:nrow(deaMonocle_tot),]
deaMonocle_tot <- merge.data.frame(deaMonocle_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaMonocle_tot$value <- as.numeric(deaMonocle_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultMonocle <- roc(deaMonocle_tot$value, 1 - deaMonocle_tot$marker_test_p_value)
# Plot the ROC curve
#plot(roc_resultMonocle)ROC from ScamPy
deaScamPy_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaScamPy <- read.csv(file.path(dirOut,paste0(file.code,"ScanPy_DEA_genes.csv")),
row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaScamPy[deaScamPy$clusters == paste0("cl",cl.val),"clusters"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
deaScamPy$data_set <- subset.datasets_csv[ind,1]
deaScamPy$set_number <- ind
deaScamPy_tot <- rbind(deaScamPy_tot, deaScamPy)
}
deaScamPy_tot <- deaScamPy_tot[2:nrow(deaScamPy_tot),]
deaScamPy_tot <- merge.data.frame(deaScamPy_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaScamPy_tot$value <- as.numeric(deaScamPy_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultScamPy <- roc(deaScamPy_tot$value, 1 - deaScamPy_tot$pval)
# Plot the ROC curve
#plot(roc_resultScamPy)Summary ROC for all methods
ThreeClusters_even <- ggroc(list(COTAN=roc_resultCOTAN, Seurat=roc_resultSeurat,
Seurat_scTr = roc_resultSeurat_scTr, Seurat_Bimod = roc_resultSeurat_Bimod,
Monocle=roc_resultMonocle, ScamPy=roc_resultScamPy))
ThreeClusters_evenPL <- ThreeClusters_even + xlab("FPR") + ylab("TPR")
ThreeClusters_evenPL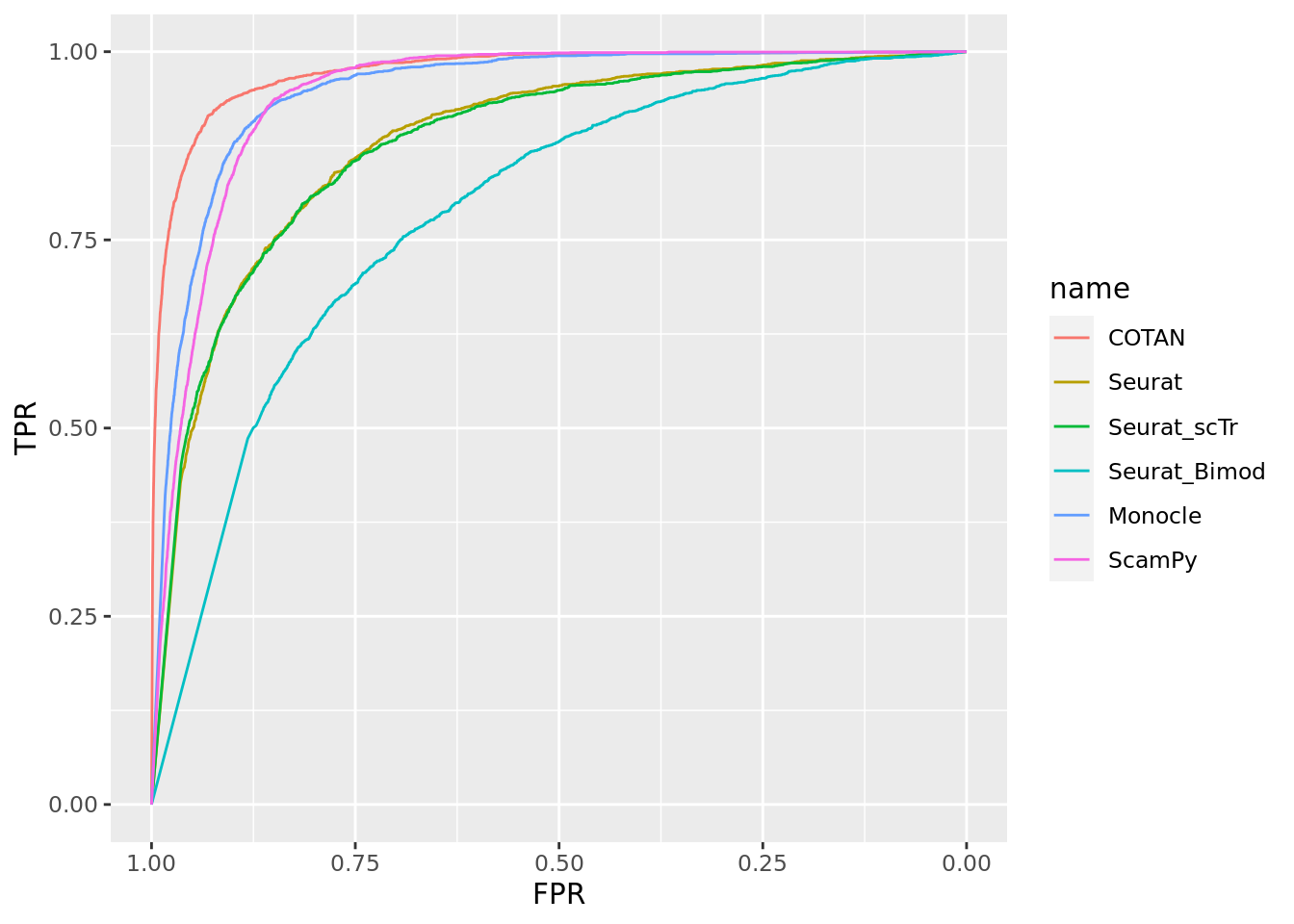
3_Clusters_uneven
True vector
subset.datasets_csv <-datasets_csv[datasets_csv$Group == "3_Clusters_uneven", ]
ground_truth_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
clusters <- str_split(subset.datasets_csv$Collection[ind], pattern = "_[+]_", simplify = T)
reads.LM.subset <- readsLogMeansPerCluster[, clusters]
ground_truth <- as.data.frame(matrix(nrow = nrow(reads.LM.subset),
ncol = ncol(reads.LM.subset)))
rownames(ground_truth) <- rownames(reads.LM.subset)
colnames(ground_truth) <- colnames(reads.LM.subset)
for (col in 1:ncol(ground_truth)) {
# log fold change
ground_truth[, col] <- reads.LM.subset[, col] - rowMeans(reads.LM.subset[, -col, drop = FALSE])
ground_truth[, col] <- ground_truth[, col] > thresholdLFC & reads.LM.subset[, col] > thresholdLFM
}
ground_truth$genes <- rownames(ground_truth)
ground_truth <- pivot_longer(ground_truth,
cols = 1:(ncol(ground_truth)-1),
names_to = "clusters")
ground_truth$data_set <- subset.datasets_csv[ind, 1]
ground_truth$set_number <- ind
ground_truth_tot <- rbind(ground_truth_tot, ground_truth)
}
ground_truth_tot <- ground_truth_tot[2:nrow(ground_truth_tot),]
head(ground_truth_tot)# A tibble: 6 × 5
genes clusters value data_set set_number
<chr> <chr> <lgl> <chr> <int>
1 Neil2 E15.0-428 FALSE 3_Clusters_uneven 1
2 Neil2 E13.5-434 FALSE 3_Clusters_uneven 1
3 Neil2 E15.0-510 FALSE 3_Clusters_uneven 1
4 Lamc1 E15.0-428 FALSE 3_Clusters_uneven 1
5 Lamc1 E13.5-434 FALSE 3_Clusters_uneven 1
6 Lamc1 E15.0-510 FALSE 3_Clusters_uneven 1length(unique(ground_truth_tot$genes))[1] 14695sum(ground_truth_tot$value)[1] 3293ROC for COTAN
onlyPositive.pVal.Cotan_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",
subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
deaCOTAN <- getClusterizationData(dataset,clName = "mergedClusters")[[2]]
pvalCOTAN <- pValueFromDEA(deaCOTAN,
numCells = getNumCells(dataset),adjustmentMethod = "none")
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.names <- c(cl.names,
str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1])
cl.names <- cl.names[!is.na(cl.names)]
}
colnames(deaCOTAN) <- cl.names
colnames(pvalCOTAN) <- cl.names
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
onlyPositive.pVal.Cotan <- pvalCOTAN
for (cl in cl.names) {
print(cl)
#temp.DEA.CotanSign <- deaCOTAN[rownames(pvalCOTAN[pvalCOTAN[,cl] < 0.05,]) ,]
onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl] <- 1 #onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl]+1
}
onlyPositive.pVal.Cotan$genes <- rownames(onlyPositive.pVal.Cotan)
onlyPositive.pVal.Cotan <- pivot_longer(onlyPositive.pVal.Cotan,
values_to = "p_values",
cols = 1:(ncol(onlyPositive.pVal.Cotan)-1),
names_to = "clusters")
onlyPositive.pVal.Cotan$data_set <- subset.datasets_csv[ind,1]
onlyPositive.pVal.Cotan$set_number <- ind
onlyPositive.pVal.Cotan_tot <- rbind(onlyPositive.pVal.Cotan_tot, onlyPositive.pVal.Cotan)
}[1] "E15.0-510"
[1] "E13.5-434"
[1] "E15.0-428"
[1] "E15.0-432"
[1] "E13.5-432"
[1] "E13.5-187"
[1] "E15.0-509"
[1] "E15.0-508"
[1] "E13.5-184"onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[2:nrow(onlyPositive.pVal.Cotan_tot),]
onlyPositive.pVal.Cotan_tot <- merge.data.frame(onlyPositive.pVal.Cotan_tot,
ground_truth_tot,by = c("genes","clusters","data_set","set_number"),all.x = T,all.y = F)
#onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[order(onlyPositive.pVal.Cotan_tot$p_values,
# decreasing = F),]
# df <- as.data.frame(matrix(nrow = nrow(onlyPositive.pVal.Cotan_tot),ncol = 3))
# colnames(df) <- c("TPR","FPR","Method")
# df$Method <- "COTAN"
#
# Positive <- sum(onlyPositive.pVal.Cotan_tot$value)
# Negative <- sum(!onlyPositive.pVal.Cotan_tot$value)
#
# for (i in 1:nrow(onlyPositive.pVal.Cotan_tot)) {
# df[i,"TPR"] <- sum(onlyPositive.pVal.Cotan_tot[1:i,"value"])/Positive
# df[i,"FPR"] <- (i-sum(onlyPositive.pVal.Cotan_tot[1:i,"value"]))/Negative
# Convert TRUE/FALSE to 1/0
onlyPositive.pVal.Cotan_tot$value <- as.numeric(onlyPositive.pVal.Cotan_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultCOTAN <- roc(onlyPositive.pVal.Cotan_tot$value, 1 - onlyPositive.pVal.Cotan_tot$p_values)
# Plot the ROC curve
#plot(roc_resultCOTAN)ROC for Seurat
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat scTransform
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_ScTransform_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_scTr <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat Bimod
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_Bimod_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_Bimod <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Monocle
deaMonocle_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaMonocle <- read.csv(file.path(dirOut,paste0(file.code,"Monocle_DEA_genes.csv")),row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaMonocle[deaMonocle$cell_group == cl.val,"cell_group"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "cell_group",
replacement = "clusters")
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "gene_id",
replacement = "genes")
deaMonocle$data_set <- subset.datasets_csv[ind,1]
deaMonocle$set_number <- ind
deaMonocle <- as.data.frame(deaMonocle)
deaMonocle_tot <- rbind(deaMonocle_tot, deaMonocle)
}
deaMonocle_tot <- deaMonocle_tot[2:nrow(deaMonocle_tot),]
deaMonocle_tot <- merge.data.frame(deaMonocle_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaMonocle_tot$value <- as.numeric(deaMonocle_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultMonocle <- roc(deaMonocle_tot$value, 1 - deaMonocle_tot$marker_test_p_value)
# Plot the ROC curve
#plot(roc_resultMonocle)ROC from ScamPy
deaScamPy_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaScamPy <- read.csv(file.path(dirOut,paste0(file.code,"ScanPy_DEA_genes.csv")),
row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaScamPy[deaScamPy$clusters == paste0("cl",cl.val),"clusters"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
deaScamPy$data_set <- subset.datasets_csv[ind,1]
deaScamPy$set_number <- ind
deaScamPy_tot <- rbind(deaScamPy_tot, deaScamPy)
}
deaScamPy_tot <- deaScamPy_tot[2:nrow(deaScamPy_tot),]
deaScamPy_tot <- merge.data.frame(deaScamPy_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaScamPy_tot$value <- as.numeric(deaScamPy_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultScamPy <- roc(deaScamPy_tot$value, 1 - deaScamPy_tot$pval)
# Plot the ROC curve
#plot(roc_resultScamPy)Summary ROC for all methods
ThreeClusters_uneven <- ggroc(list(COTAN=roc_resultCOTAN, Seurat=roc_resultSeurat,
Seurat_scTr = roc_resultSeurat_scTr, Seurat_Bimod = roc_resultSeurat_Bimod,
Monocle=roc_resultMonocle, ScamPy=roc_resultScamPy))
ThreeClusters_unevenPL <- ThreeClusters_uneven + xlab("FPR") + ylab("TPR")
ThreeClusters_unevenPL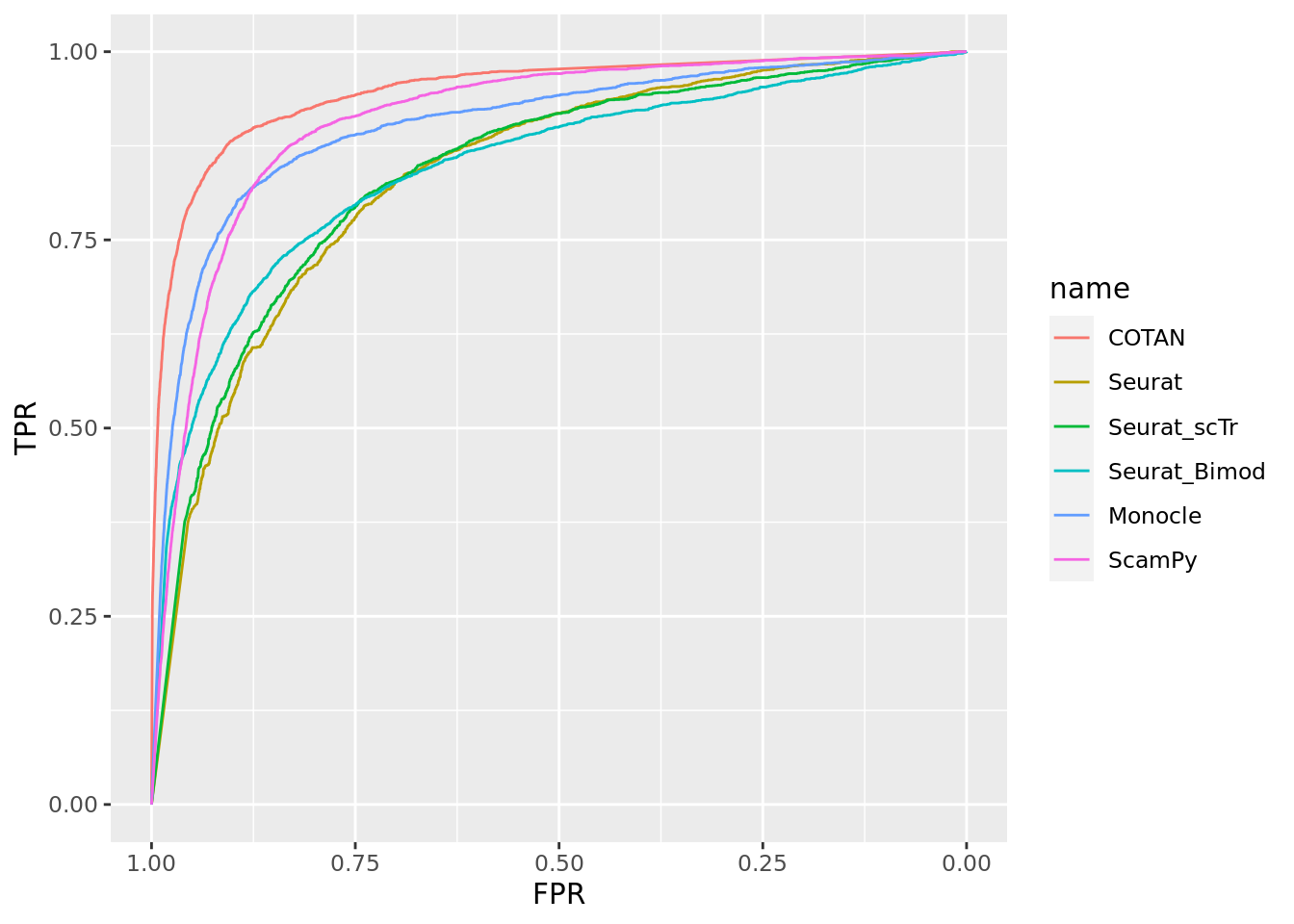
5 clusters
5_Clusters_uneven
True vector
subset.datasets_csv <-datasets_csv[datasets_csv$Group == "5_Clusters_uneven", ]
ground_truth_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
clusters <- str_split(subset.datasets_csv$Collection[ind], pattern = "_[+]_", simplify = T)
reads.LM.subset <- readsLogMeansPerCluster[, clusters]
ground_truth <- as.data.frame(matrix(nrow = nrow(reads.LM.subset),
ncol = ncol(reads.LM.subset)))
rownames(ground_truth) <- rownames(reads.LM.subset)
colnames(ground_truth) <- colnames(reads.LM.subset)
for (col in 1:ncol(ground_truth)) {
# log fold change
ground_truth[, col] <- reads.LM.subset[, col] - rowMeans(reads.LM.subset[, -col, drop = FALSE])
ground_truth[, col] <- ground_truth[, col] > thresholdLFC & reads.LM.subset[, col] > thresholdLFM
}
ground_truth$genes <- rownames(ground_truth)
ground_truth <- pivot_longer(ground_truth,
cols = 1:(ncol(ground_truth)-1),
names_to = "clusters")
ground_truth$data_set <- subset.datasets_csv[ind, 1]
ground_truth$set_number <- ind
ground_truth_tot <- rbind(ground_truth_tot, ground_truth)
}
ground_truth_tot <- ground_truth_tot[2:nrow(ground_truth_tot),]
head(ground_truth_tot)# A tibble: 6 × 5
genes clusters value data_set set_number
<chr> <chr> <lgl> <chr> <int>
1 Neil2 E13.5-510 FALSE 5_Clusters_uneven 1
2 Neil2 E15.0-437 FALSE 5_Clusters_uneven 1
3 Neil2 E15.0-510 FALSE 5_Clusters_uneven 1
4 Neil2 E13.5-432 FALSE 5_Clusters_uneven 1
5 Neil2 E13.5-437 FALSE 5_Clusters_uneven 1
6 Lamc1 E13.5-510 FALSE 5_Clusters_uneven 1length(unique(ground_truth_tot$genes))[1] 14695sum(ground_truth_tot$value)[1] 3653ROC for COTAN
onlyPositive.pVal.Cotan_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",
subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
deaCOTAN <- getClusterizationData(dataset,clName = "mergedClusters")[[2]]
pvalCOTAN <- pValueFromDEA(deaCOTAN,
numCells = getNumCells(dataset),adjustmentMethod = "none")
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.names <- c(cl.names,
str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1])
cl.names <- cl.names[!is.na(cl.names)]
}
colnames(deaCOTAN) <- cl.names
colnames(pvalCOTAN) <- cl.names
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
onlyPositive.pVal.Cotan <- pvalCOTAN
for (cl in cl.names) {
print(cl)
#temp.DEA.CotanSign <- deaCOTAN[rownames(pvalCOTAN[pvalCOTAN[,cl] < 0.05,]) ,]
onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl] <- 1 #onlyPositive.pVal.Cotan[rownames(deaCOTAN[deaCOTAN[,cl] < 0,]),cl]+1
}
onlyPositive.pVal.Cotan$genes <- rownames(onlyPositive.pVal.Cotan)
onlyPositive.pVal.Cotan <- pivot_longer(onlyPositive.pVal.Cotan,
values_to = "p_values",
cols = 1:(ncol(onlyPositive.pVal.Cotan)-1),
names_to = "clusters")
onlyPositive.pVal.Cotan$data_set <- subset.datasets_csv[ind,1]
onlyPositive.pVal.Cotan$set_number <- ind
onlyPositive.pVal.Cotan_tot <- rbind(onlyPositive.pVal.Cotan_tot, onlyPositive.pVal.Cotan)
}[1] "E13.5-432"
[1] "E15.0-510"
[1] "E13.5-437"
[1] "E15.0-437"
[1] "E13.5-510"
[1] "E13.5-434"
[1] "E15.0-428"
[1] "E13.5-184"
[1] "E15.0-434"
[1] "E17.5-505"
[1] "E15.0-432"
[1] "E13.5-432"
[1] "E15.0-509"
[1] "E15.0-508"
[1] "E13.5-187"onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[2:nrow(onlyPositive.pVal.Cotan_tot),]
onlyPositive.pVal.Cotan_tot <- merge.data.frame(onlyPositive.pVal.Cotan_tot,
ground_truth_tot,by = c("genes","clusters","data_set","set_number"),all.x = T,all.y = F)
#onlyPositive.pVal.Cotan_tot <- onlyPositive.pVal.Cotan_tot[order(onlyPositive.pVal.Cotan_tot$p_values,
# decreasing = F),]
# df <- as.data.frame(matrix(nrow = nrow(onlyPositive.pVal.Cotan_tot),ncol = 3))
# colnames(df) <- c("TPR","FPR","Method")
# df$Method <- "COTAN"
#
# Positive <- sum(onlyPositive.pVal.Cotan_tot$value)
# Negative <- sum(!onlyPositive.pVal.Cotan_tot$value)
#
# for (i in 1:nrow(onlyPositive.pVal.Cotan_tot)) {
# df[i,"TPR"] <- sum(onlyPositive.pVal.Cotan_tot[1:i,"value"])/Positive
# df[i,"FPR"] <- (i-sum(onlyPositive.pVal.Cotan_tot[1:i,"value"]))/Negative
# Convert TRUE/FALSE to 1/0
onlyPositive.pVal.Cotan_tot$value <- as.numeric(onlyPositive.pVal.Cotan_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultCOTAN <- roc(onlyPositive.pVal.Cotan_tot$value, 1 - onlyPositive.pVal.Cotan_tot$p_values)
# Plot the ROC curve
#plot(roc_resultCOTAN)ROC for Seurat
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat scTransform
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_ScTransform_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_scTr <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Seurat Bimod
deaSeurat_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
#print(ind)
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
deaSeurat <- read.csv(file.path(dirOut,paste0(file.code,"Seurat_DEA_Bimod_genes.csv")), row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaSeurat[deaSeurat$cluster == cl.val,]$cluster <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "cluster",
replacement = "clusters")
colnames(deaSeurat) <- str_replace(colnames(deaSeurat),
pattern = "gene",
replacement = "genes")
deaSeurat$data_set <- subset.datasets_csv[ind,1]
deaSeurat$set_number <- ind
deaSeurat_tot <- rbind(deaSeurat_tot, deaSeurat)
}
deaSeurat_tot <- deaSeurat_tot[2:nrow(deaSeurat_tot),]
deaSeurat_tot <- merge.data.frame(deaSeurat_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaSeurat_tot$value <- as.numeric(deaSeurat_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultSeurat_Bimod <- roc(deaSeurat_tot$value, 1 - deaSeurat_tot$p_val)
# Plot the ROC curve
#plot(roc_resultSeurat)ROC for Monocle
deaMonocle_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaMonocle <- read.csv(file.path(dirOut,paste0(file.code,"Monocle_DEA_genes.csv")),row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaMonocle[deaMonocle$cell_group == cl.val,"cell_group"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "cell_group",
replacement = "clusters")
colnames(deaMonocle) <- str_replace(colnames(deaMonocle),
pattern = "gene_id",
replacement = "genes")
deaMonocle$data_set <- subset.datasets_csv[ind,1]
deaMonocle$set_number <- ind
deaMonocle <- as.data.frame(deaMonocle)
deaMonocle_tot <- rbind(deaMonocle_tot, deaMonocle)
}
deaMonocle_tot <- deaMonocle_tot[2:nrow(deaMonocle_tot),]
deaMonocle_tot <- merge.data.frame(deaMonocle_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaMonocle_tot$value <- as.numeric(deaMonocle_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultMonocle <- roc(deaMonocle_tot$value, 1 - deaMonocle_tot$marker_test_p_value)
# Plot the ROC curve
#plot(roc_resultMonocle)ROC from ScamPy
deaScamPy_tot <- NA
for (ind in 1:dim(subset.datasets_csv)[1]) {
file.code <- paste0(subset.datasets_csv$Group[ind],"_",subset.datasets_csv$Collection[ind])
dataset <- readRDS(file = file.path(dataSetDir,paste0(file.code,".RDS")))
clusterization <- getClusterizationData(dataset, clName = "mergedClusters")[[1]]
#print(file.code)
deaScamPy <- read.csv(file.path(dirOut,paste0(file.code,"ScanPy_DEA_genes.csv")),
row.names = 1)
cl.names <- NA
for (cl.val in unique(clusterization)) {
#print(cl.val)
cl.name <- str_split(names(clusterization[clusterization == cl.val])[1],
pattern = "_",simplify = T)[1]
cl.names <- c(cl.names,cl.name)
cl.names <- cl.names[!is.na(cl.names)]
deaScamPy[deaScamPy$clusters == paste0("cl",cl.val),"clusters"] <- cl.name
}
clusters <- str_split(datasets_csv$Collection[ind],pattern = "_[+]_",simplify = T)
deaScamPy$data_set <- subset.datasets_csv[ind,1]
deaScamPy$set_number <- ind
deaScamPy_tot <- rbind(deaScamPy_tot, deaScamPy)
}
deaScamPy_tot <- deaScamPy_tot[2:nrow(deaScamPy_tot),]
deaScamPy_tot <- merge.data.frame(deaScamPy_tot,
ground_truth_tot,
by = c("genes","clusters","data_set","set_number"),
all.x = T,all.y = F)
# Convert TRUE/FALSE to 1/0
deaScamPy_tot$value <- as.numeric(deaScamPy_tot$value)
# Compute the ROC curve - note that we invert the p-values with 1 - p_values
roc_resultScamPy <- roc(deaScamPy_tot$value, 1 - deaScamPy_tot$pval)
# Plot the ROC curve
#plot(roc_resultScamPy)Summary ROC for all methods
FiveClusters_uneven <- ggroc(list(COTAN=roc_resultCOTAN, Seurat=roc_resultSeurat,
Seurat_scTr = roc_resultSeurat_scTr, Seurat_Bimod = roc_resultSeurat_Bimod,
Monocle=roc_resultMonocle, ScamPy=roc_resultScamPy))
FiveClusters_unevenPL <- FiveClusters_uneven + xlab("FPR") + ylab("TPR")
FiveClusters_unevenPL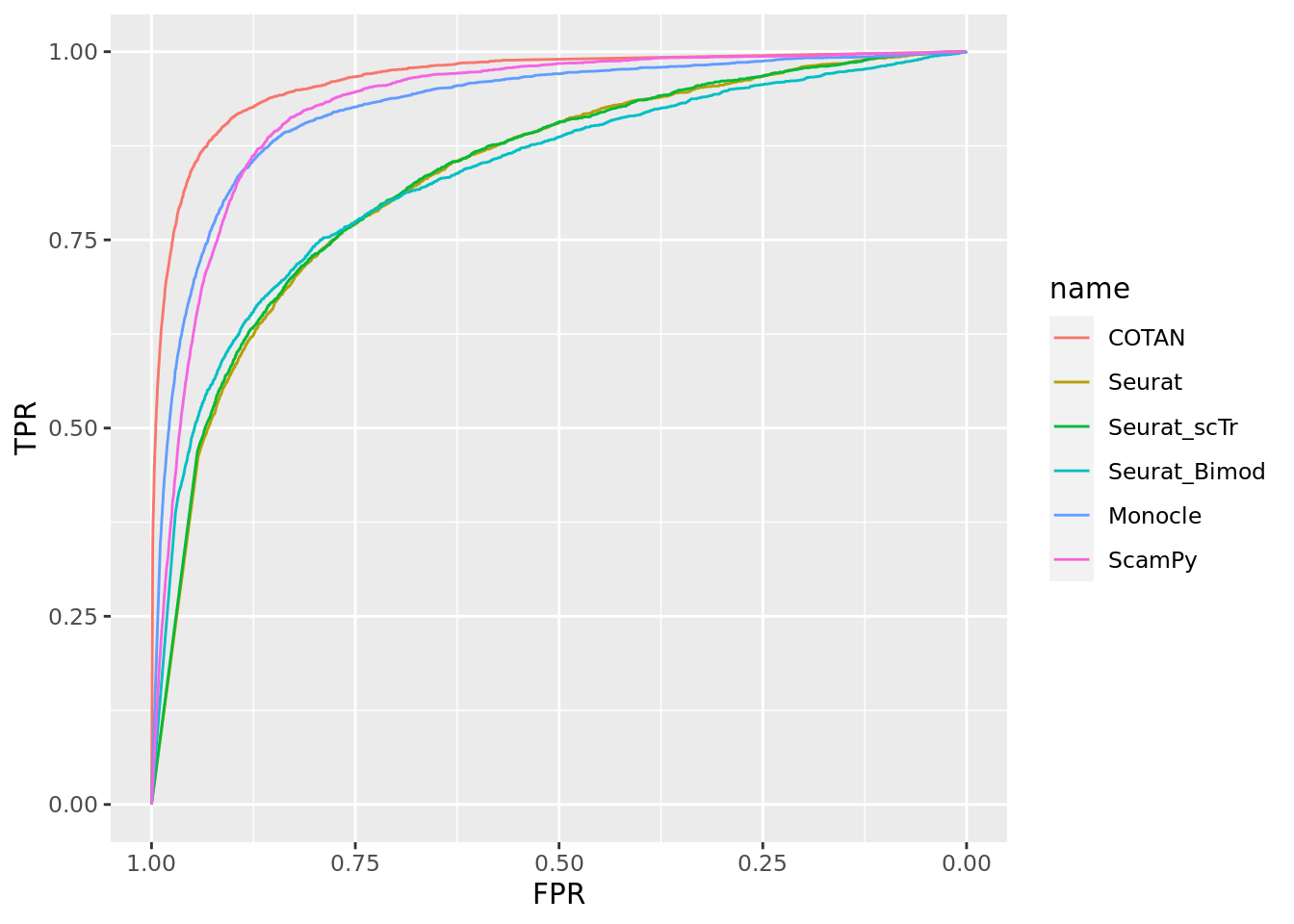
Global Summary
2 Clusters
ggarrange(TwoClusters_even_nearPL,TwoClusters_even_mediumPL, TwoClusters_even_farPL,TwoClusters_uneven_nearPL, TwoClusters_uneven_mediumPL,TwoClusters_uneven_farPL,
labels = c("Even_Near", "Even_Medium", "Even_Far", "Uneven_Near","Uneven_Medium","Uneven_Far"),
ncol = 3, nrow = 2, common.legend = T, legend = "bottom")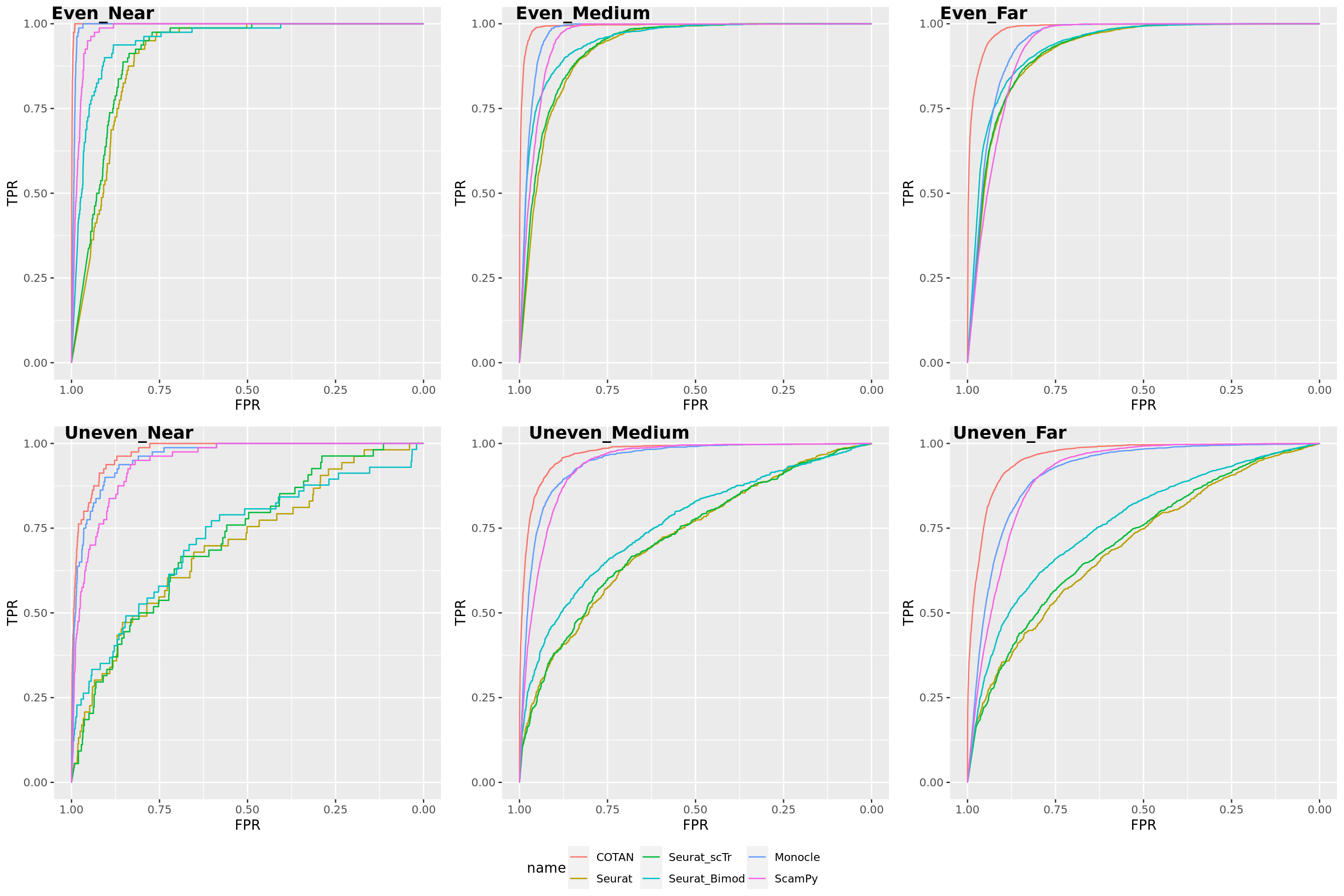
3 and 5 Clusters
ggarrange(ThreeClusters_evenPL,ThreeClusters_unevenPL, NULL, FiveClusters_unevenPL,
labels = c("3_Even", "3_Uneven", "", "5_Uneven"),
ncol = 2, nrow = 2, common.legend = T, legend = "bottom")